Tabb David L, Kaniyar Mohammed Hanzala, Bringas Omar G Rosas, Shin Heaji, Di Stefano Luciano, Taylor Martin S, Xie Shaoshuai, Yilmaz Omer H, LaCava John
European Research Institute for the Biology of Ageing, University Medical Center Groningen, Groningen, The Netherlands.
Department of Biology, David H. Koch Institute for Integrative Cancer Research at MIT, MIT, Cambridge, MA USA.
J Proteins Proteom. 2024;15(3):281-298. doi: 10.1007/s42485-024-00166-4. Epub 2024 Sep 17.
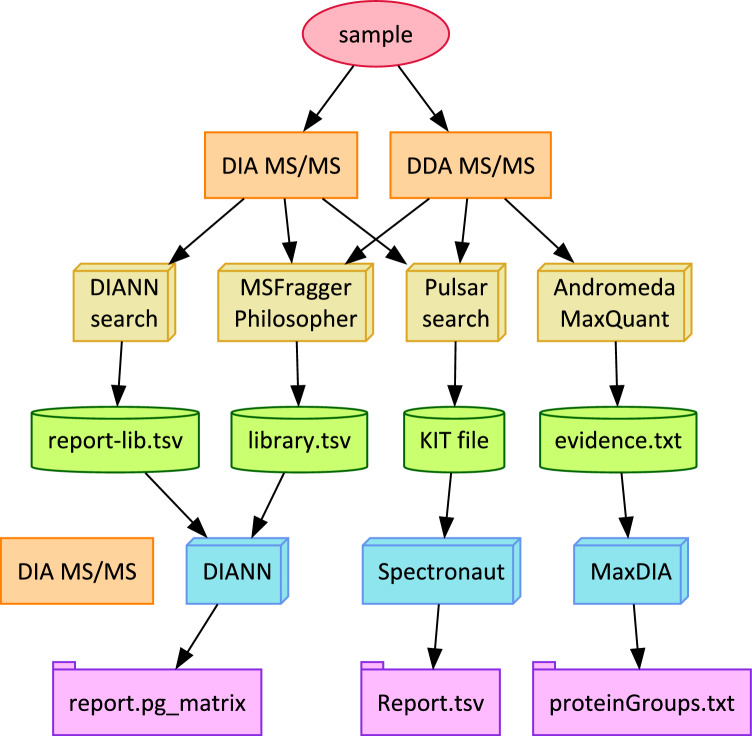
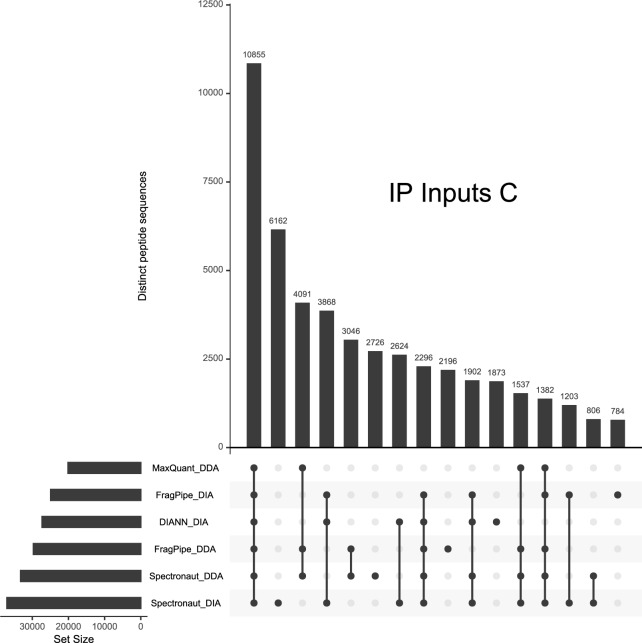
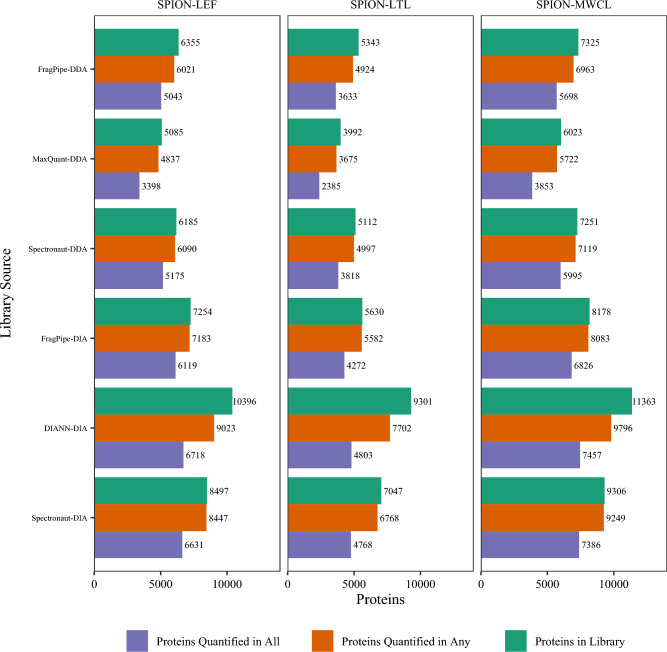
Data-Independent Acquisition (DIA) LC-MS/MS is an attractive partner for co-immunoprecipitation (co-IP) and affinity proteomics in general. Reducing the variability of quantitation by DIA could increase the statistical contrast for detecting specific interactors versus what has been achieved in Data-Dependent Acquisition (DDA). By interrogating affinity proteomes featuring both DDA and DIA experiments, we sought to evaluate the spectral libraries, the missingness of protein quantity tables, and the CV of protein quantities in six studies representing three different instrument manufacturers. We examined four contemporary bioinformatics workflows for DIA: FragPipe, DIA-NN, Spectronaut, and MaxQuant. We determined that (1) identifying spectral libraries directly from DIA experiments works well enough that separate DDA experiments do not produce larger spectral libraries when given equivalent instrument time; (2) experiments involving mock pull-downs or IgG controls may feature such indistinct signals that contemporary software will struggle to quantify them; (3) measured CV values were well controlled by Spectronaut and DIA-NN (and FragPipe, which implements DIA-NN for the quantitation step); and (4) when FragPipe builds spectral libraries and quantifies proteins from DIA experiments rather than performing both operations in DDA experiments, the DIA route results in a larger number of proteins quantified without missing values as well as lower CV for measured protein quantities.
The online version contains supplementary material available at 10.1007/s42485-024-00166-4.
数据非依赖型采集(DIA)液相色谱-串联质谱(LC-MS/MS)总体而言是免疫共沉淀(co-IP)和亲和蛋白质组学的理想搭档。通过减少DIA定量的变异性,与数据依赖型采集(DDA)相比,检测特定相互作用分子时的统计对比度可能会增加。通过对同时包含DDA和DIA实验的亲和蛋白质组进行分析,我们试图评估来自三家不同仪器制造商的六项研究中的谱库、蛋白质定量表的缺失情况以及蛋白质定量的变异系数(CV)。我们研究了四种用于DIA的当代生物信息学工作流程:FragPipe、DIA-NN、Spectronaut和MaxQuant。我们确定:(1)直接从DIA实验中识别谱库效果良好,在给予相同仪器时间的情况下,单独的DDA实验不会产生更大的谱库;(2)涉及模拟下拉或IgG对照的实验可能具有非常模糊的信号,以至于当代软件难以对其进行定量;(3)Spectronaut和DIA-NN(以及在定量步骤中采用DIA-NN的FragPipe)能很好地控制测量的CV值;(4)当FragPipe构建谱库并对DIA实验中的蛋白质进行定量,而不是在DDA实验中同时执行这两个操作时,DIA方法会导致更多无缺失值定量的蛋白质,并且测量的蛋白质定量的CV更低。
在线版本包含可在10.1007/s42485-024-00166-4获取的补充材料。