Hernández-Serda Manuel Alejandro, Vázquez-Valadez Víctor H, Aguirre-Vidal Pablo, Markarian Nathan M, Medina-Franco José L, Cardenas-Granados Luis Alfonso, Alarcón-López Aldo Yoshio, Martínez-Soriano Pablo A, Velázquez-Sánchez Ana María, Falfán-Valencia Rodolfo E, Angeles Enrique, Abrahamyan Levon
Departamento de Ciencias Químicas FES Cuautitlán, Universidad Nacional Autónoma de México (UNAM), Av. 1 de Mayo SN Cuautitlán Izcalli, Mexico City 54750, Mexico.
Departamento de Ciencias Biológicas FES Cuautitlán, Universidad Nacional Autónoma de México (UNAM), Av. 1 de Mayo SN Cuautitlán Izcalli, Mexico City 54750, Mexico.
Pathogens. 2024 Oct 11;13(10):887. doi: 10.3390/pathogens13100887.
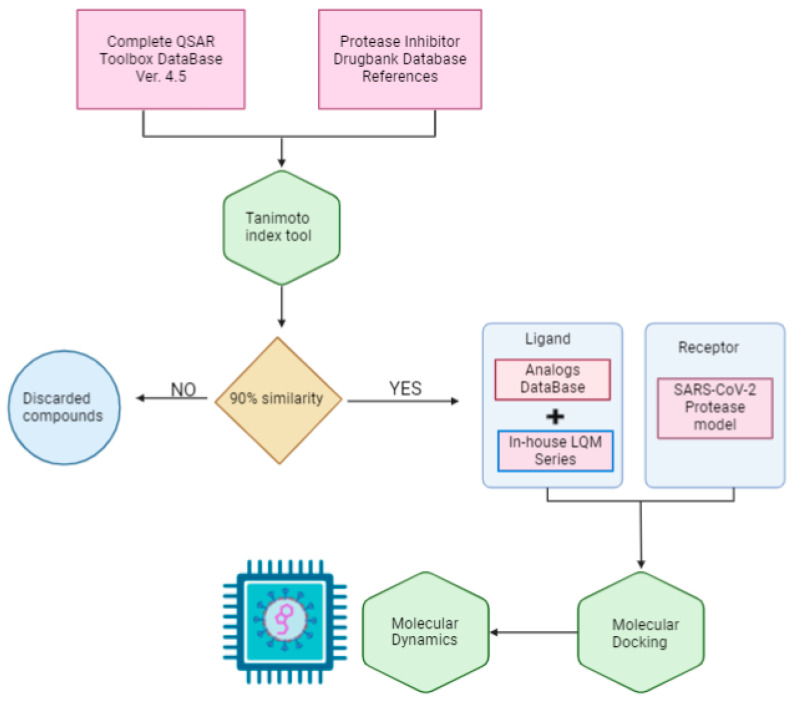
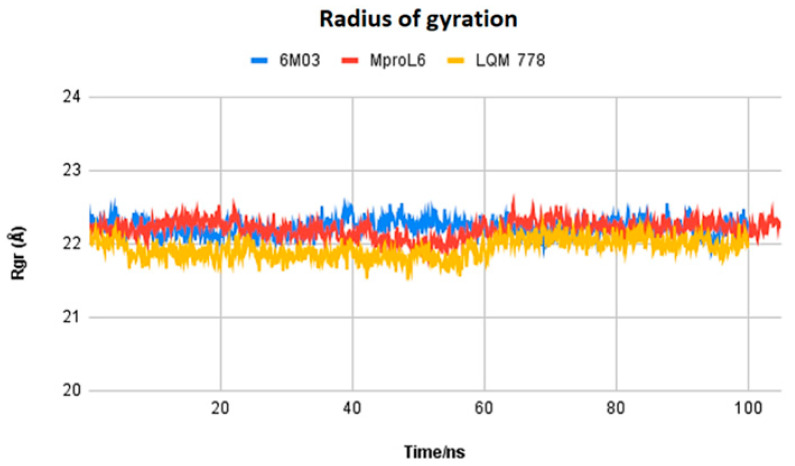
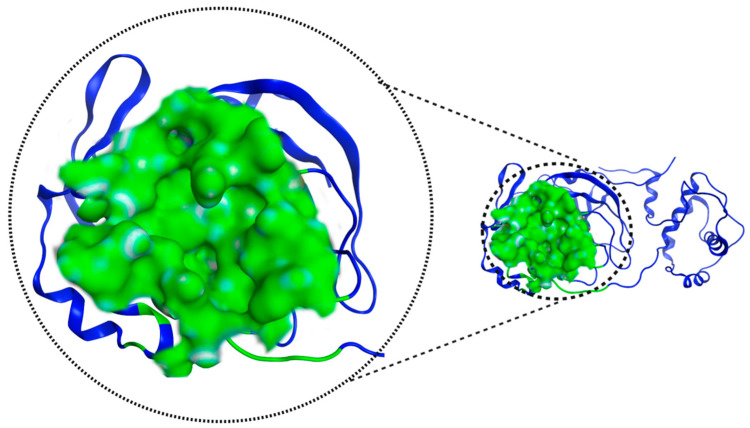
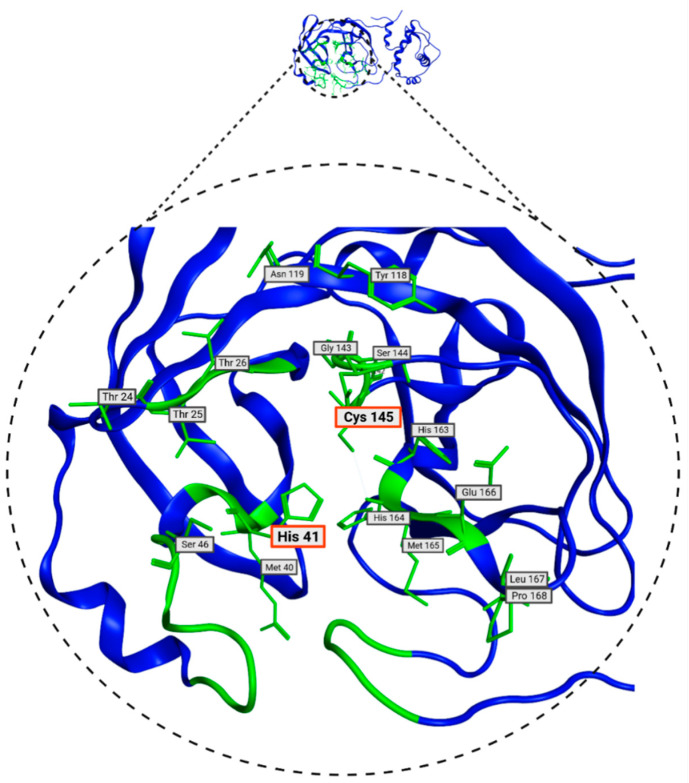
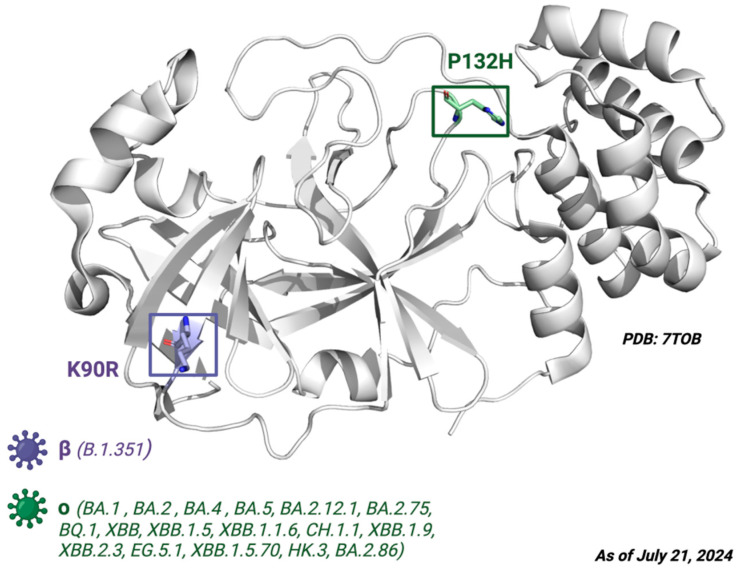
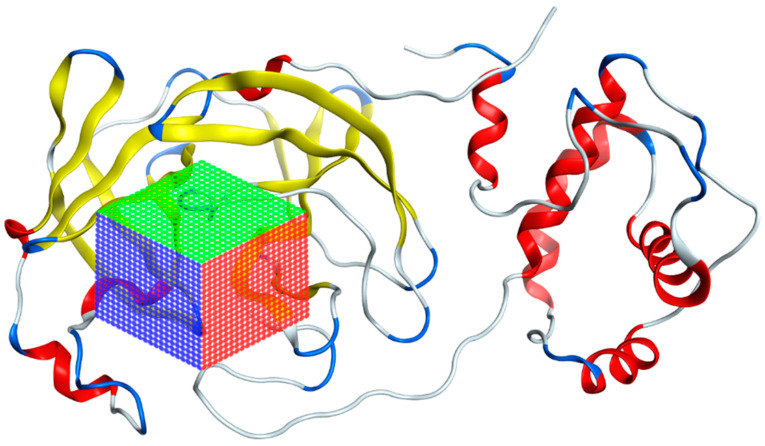
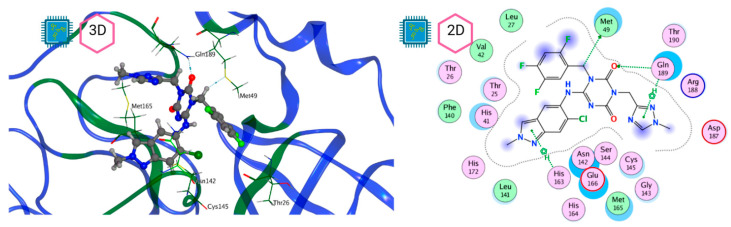
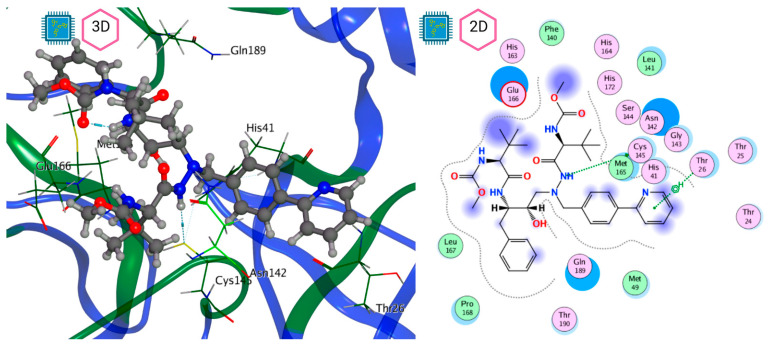
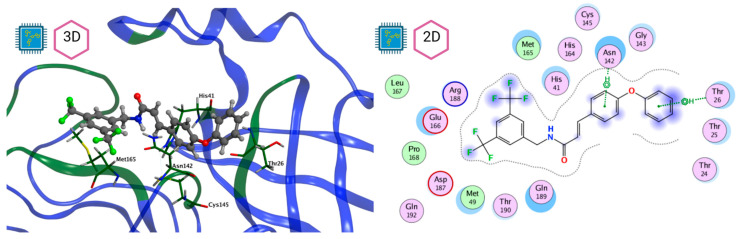
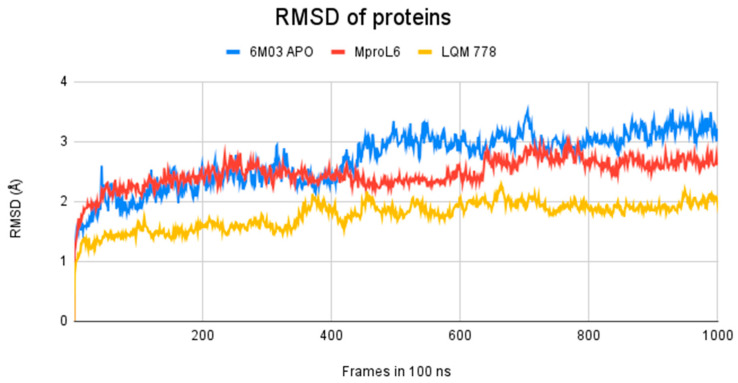
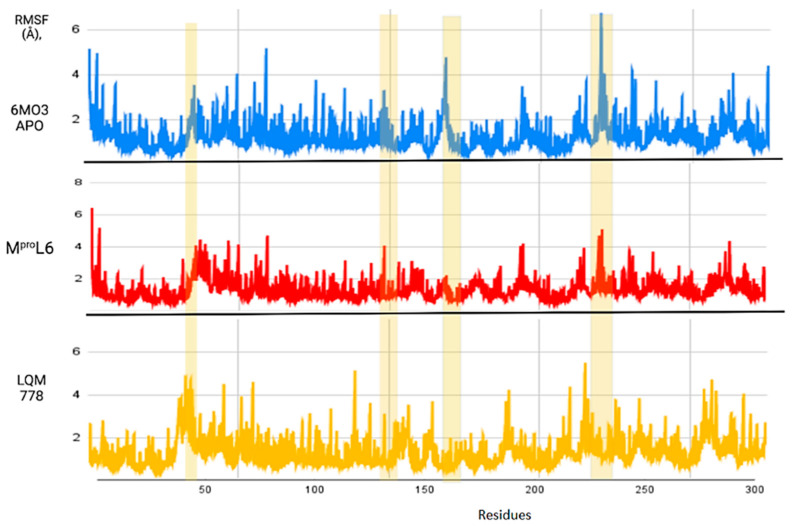
The ongoing Coronavirus Disease 19 (COVID-19) pandemic has had a profound impact on the global healthcare system. As the SARS-CoV-2 virus, responsible for this pandemic, continues to spread and develop mutations in its genetic material, new variants of interest (VOIs) and variants of concern (VOCs) are emerging. These outbreaks lead to a decrease in the efficacy of existing treatments such as vaccines or drugs, highlighting the urgency of new therapies for COVID-19. Therefore, in this study, we aimed to identify potential SARS-CoV-2 antivirals using a virtual screening protocol and molecular dynamics simulations. These techniques allowed us to predict the binding affinity of a database of compounds with the virus M protein. This in silico approach enabled us to identify twenty-two chemical structures from a public database (QSAR Toolbox Ver 4.5 ) and ten promising molecules from our in-house database. The latter molecules possess advantageous qualities, such as two-step synthesis, cost-effectiveness, and long-lasting physical and chemical stability. Consequently, these molecules can be considered as promising alternatives to combat emerging SARS-CoV-2 variants.
持续的新型冠状病毒肺炎(COVID-19)大流行对全球医疗系统产生了深远影响。随着导致此次大流行的严重急性呼吸综合征冠状病毒2(SARS-CoV-2)病毒继续传播并在其遗传物质中发生突变,新的关注变异株(VOIs)和关切变异株(VOCs)不断出现。这些疫情导致疫苗或药物等现有治疗方法的疗效下降,凸显了针对COVID-19的新疗法的紧迫性。因此,在本研究中,我们旨在使用虚拟筛选方案和分子动力学模拟来鉴定潜在的SARS-CoV-2抗病毒药物。这些技术使我们能够预测化合物数据库与病毒M蛋白的结合亲和力。这种计算机模拟方法使我们能够从公共数据库(QSAR Toolbox Ver 4.5)中识别出22种化学结构,并从我们的内部数据库中识别出10种有前景的分子。后一类分子具有诸如两步合成、成本效益以及持久的物理和化学稳定性等优势特性。因此,这些分子可被视为对抗新出现的SARS-CoV-2变异株的有前景的替代物。