Cao Piao-Yang, He Yang, Cui Ming-Yang, Zhang Xiao-Min, Zhang Qingye, Zhang Hong-Yu
Hubei Key Laboratory of Agricultural Bioinformatics, College of Informatics, Huazhong Agricultural University, Wuhan, 430070, People's Republic of China.
J Cheminform. 2024 Nov 28;16(1):133. doi: 10.1186/s13321-024-00933-x.
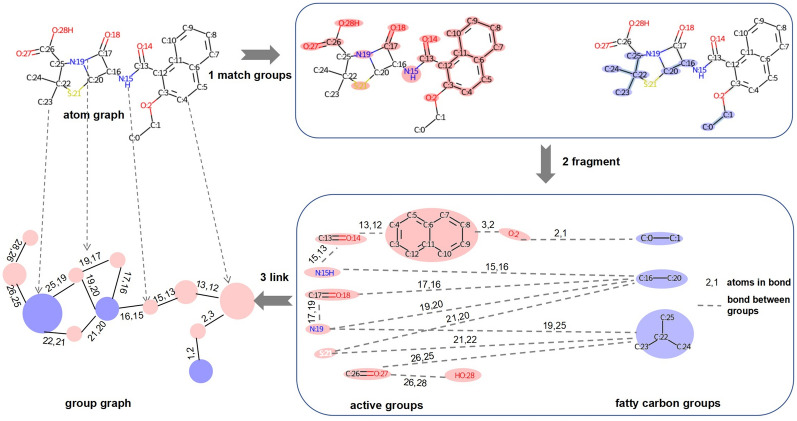
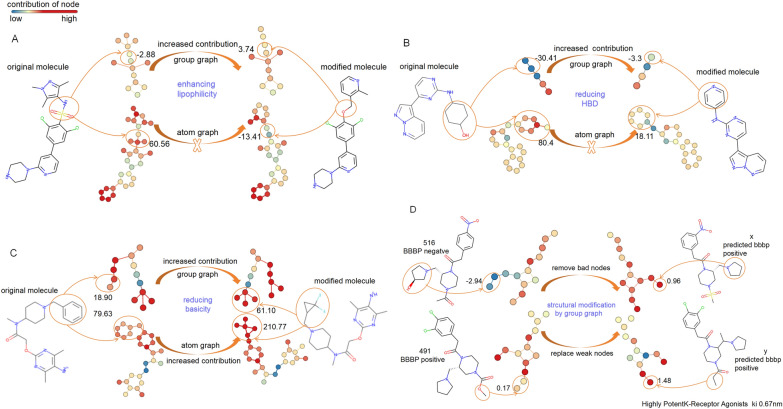
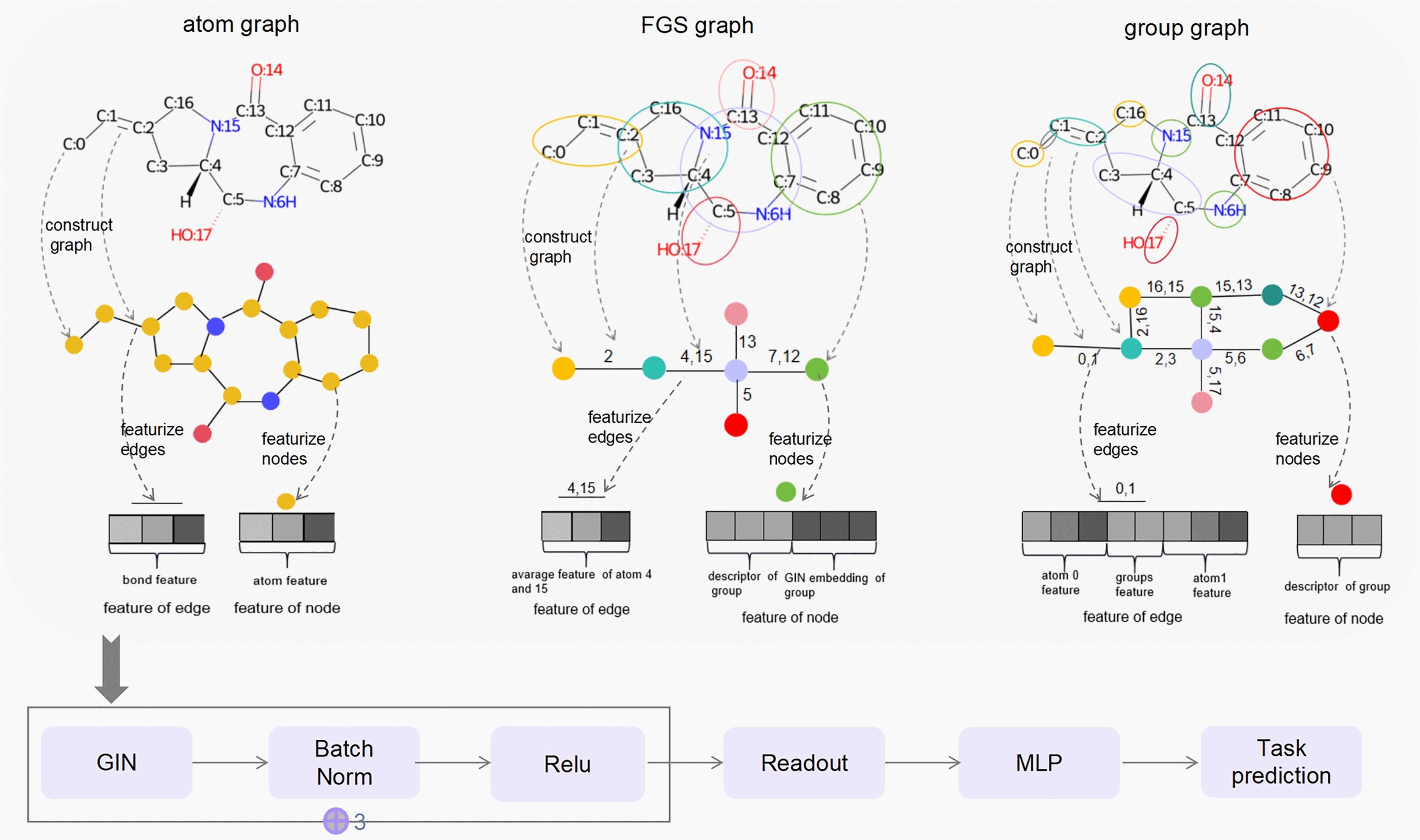
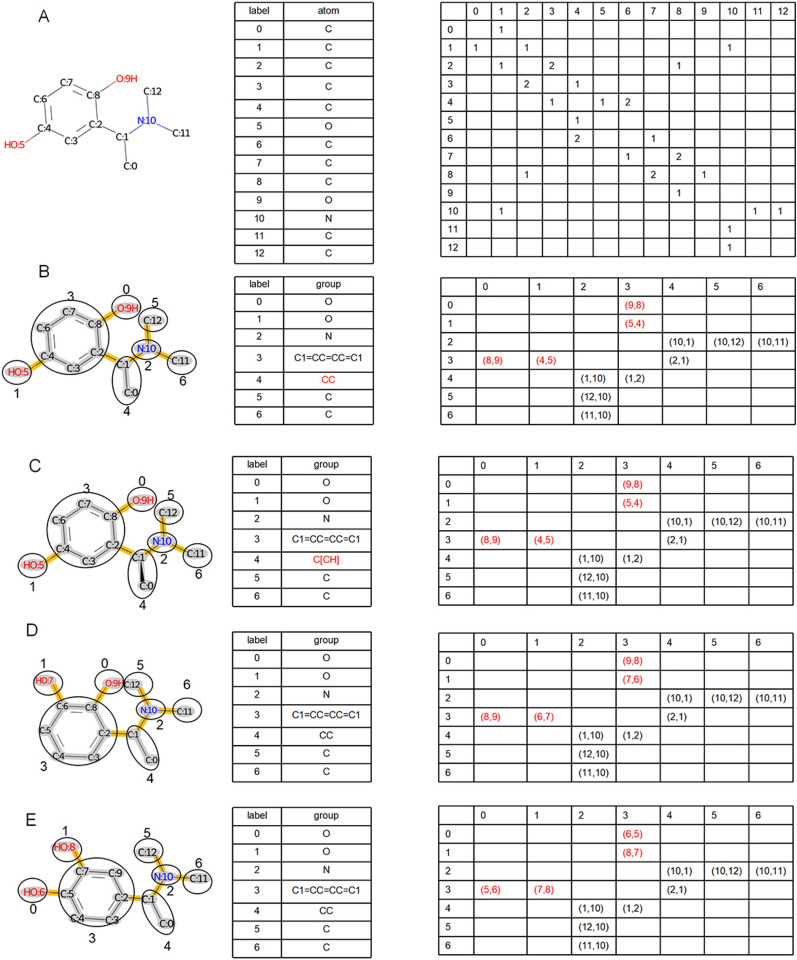
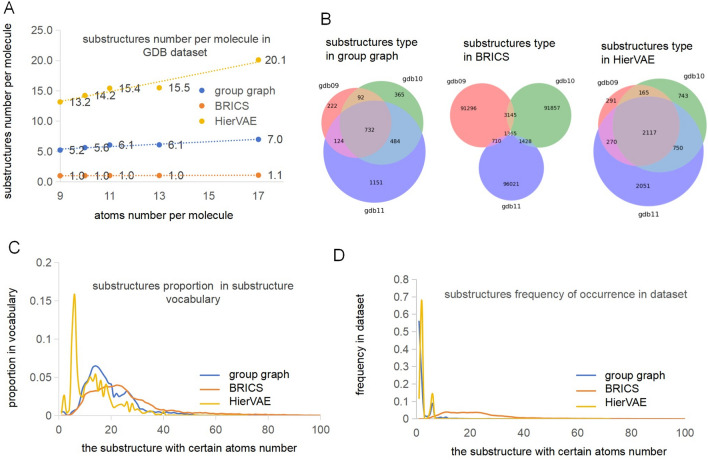
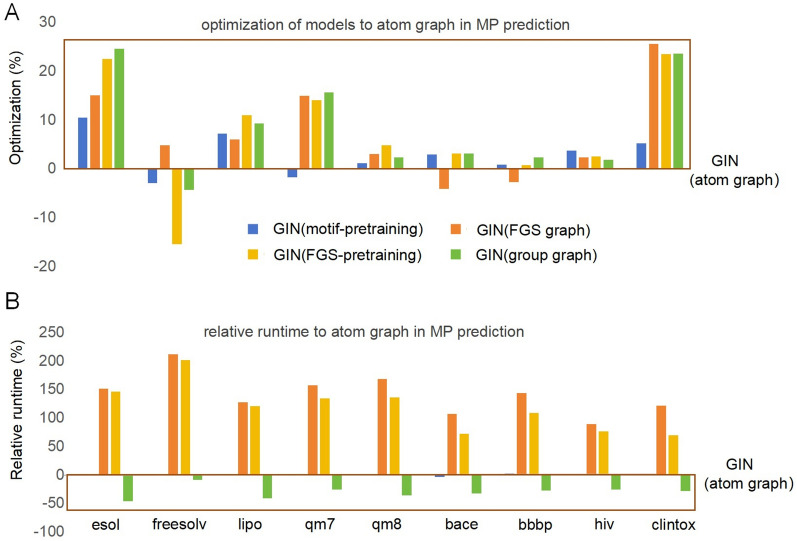
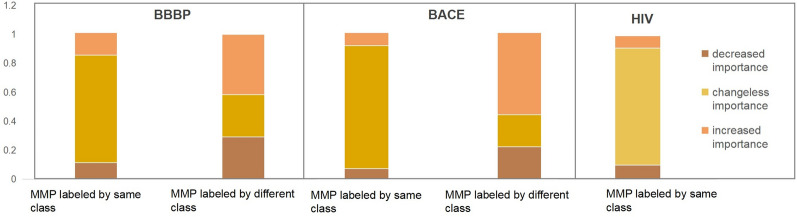
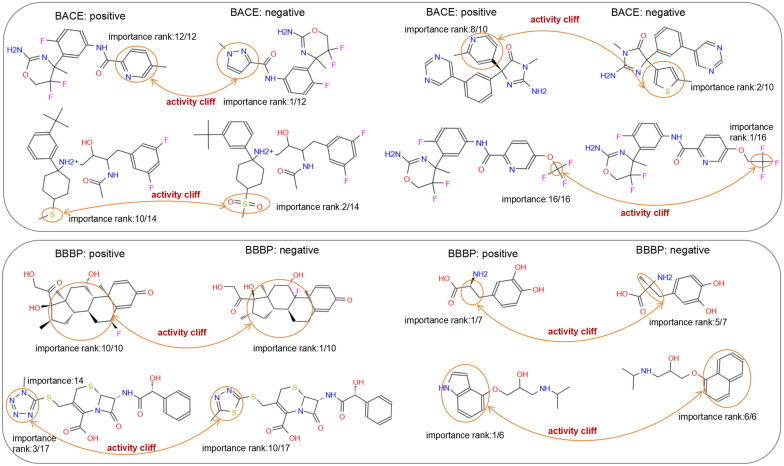
The exploration of chemical space holds promise for developing influential chemical entities. Molecular representations, which reflect features of molecular structure in silico, assist in navigating chemical space appropriately. Unlike atom-level molecular representations, such as SMILES and atom graph, which can sometimes lead to confusing interpretations about chemical substructures, substructure-level molecular representations encode important substructures into molecular features; they not only provide more information for predicting molecular properties and drug‒drug interactions but also help to interpret the correlations between molecular properties and substructures. However, it remains challenging to represent the entire molecular structure both intactly and simply with substructure-level molecular representations. In this study, we developed a novel substructure-level molecular representation and named it a group graph. The group graph offers three advantages: (a) the substructure of the group graph reflects the diversity and consistency of different molecular datasets; (b) the group graph retains molecular structural features with minimal information loss because the graph isomorphism network (GIN) of the group graph performs well in molecular properties and drug‒drug interactions prediction, showing higher accuracy and efficiency than the model of other molecular graphs, even without any pretraining; and (c) the molecular property may change when the substructure is substituted with another of differing importance in group graph, facilitating the detection of activity cliffs. In addition, we successfully predicted structural modifications to improve blood‒brain barrier permeability (BBBP) via the GIN of group graph. Therefore, the group graph takes advantages for simultaneously representing molecular local characteristics and global features.Scientific contribution The group graph, as a substructure-level molecular representation, has the ability to retain molecular structural features with minimal information loss. As a result, it shows superior performance in predicting molecular properties and drug‒drug interactions with enhanced efficiency and interpretability.
化学空间的探索为开发有影响力的化学实体带来了希望。分子表示法在计算机上反映分子结构的特征,有助于恰当地探索化学空间。与原子级分子表示法(如SMILES和原子图)不同,后者有时会导致对化学子结构的解释令人困惑,子结构级分子表示法将重要子结构编码为分子特征;它们不仅为预测分子性质和药物-药物相互作用提供了更多信息,还有助于解释分子性质与子结构之间的相关性。然而,用子结构级分子表示法完整而简单地表示整个分子结构仍然具有挑战性。在本研究中,我们开发了一种新的子结构级分子表示法,并将其命名为基团图。基团图具有三个优点:(a)基团图的子结构反映了不同分子数据集的多样性和一致性;(b)基团图以最小的信息损失保留分子结构特征,因为基团图的图同构网络(GIN)在分子性质和药物-药物相互作用预测方面表现良好,即使没有任何预训练,也比其他分子图模型显示出更高的准确性和效率;(c)在基团图中,当子结构被另一个重要性不同的子结构取代时,分子性质可能会发生变化,这有助于检测活性悬崖。此外,我们通过基团图的GIN成功预测了改善血脑屏障通透性(BBBP)的结构修饰。因此,基团图在同时表示分子局部特征和全局特征方面具有优势。科学贡献基团图作为一种子结构级分子表示法,能够以最小的信息损失保留分子结构特征。因此,它在预测分子性质和药物-药物相互作用方面表现出卓越的性能,具有更高的效率和可解释性。