Batatia Ilyes, Batzner Simon, Kovács Dávid Péter, Musaelian Albert, Simm Gregor N C, Drautz Ralf, Ortner Christoph, Kozinsky Boris, Csányi Gábor
Engineering Laboratory, University of Cambridge, Cambridge, UK.
Department of Chemistry, ENS Paris-Saclay, Université Paris-Saclay, Gif-sur-Yvette, France.
Nat Mach Intell. 2025;7(1):56-67. doi: 10.1038/s42256-024-00956-x. Epub 2025 Jan 15.
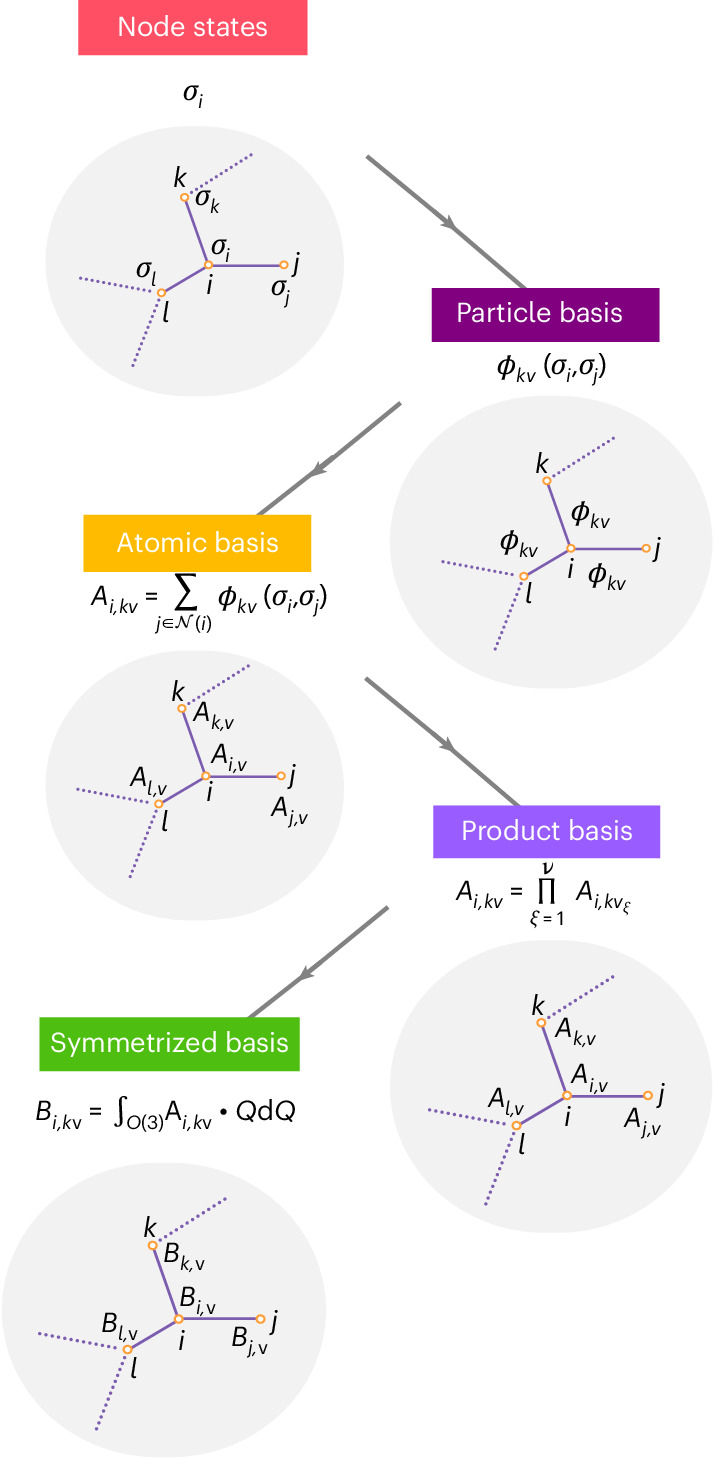
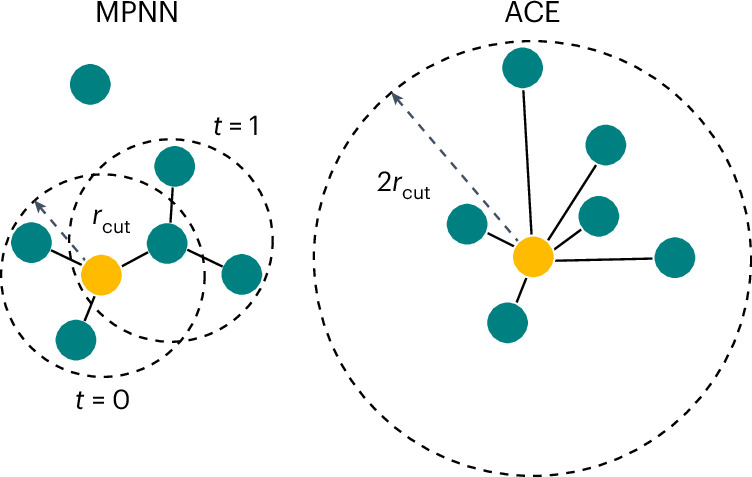
Molecular dynamics simulation is an important tool in computational materials science and chemistry, and in the past decade it has been revolutionized by machine learning. This rapid progress in machine learning interatomic potentials has produced a number of new architectures in just the past few years. Particularly notable among these are the atomic cluster expansion, which unified many of the earlier ideas around atom-density-based descriptors, and Neural Equivariant Interatomic Potentials (NequIP), a message-passing neural network with equivariant features that exhibited state-of-the-art accuracy at the time. Here we construct a mathematical framework that unifies these models: atomic cluster expansion is extended and recast as one layer of a multi-layer architecture, while the linearized version of NequIP is understood as a particular sparsification of a much larger polynomial model. Our framework also provides a practical tool for systematically probing different choices in this unified design space. An ablation study of NequIP, via a set of experiments looking at in- and out-of-domain accuracy and smooth extrapolation very far from the training data, sheds some light on which design choices are critical to achieving high accuracy. A much-simplified version of NequIP, which we call BOTnet (for body-ordered tensor network), has an interpretable architecture and maintains its accuracy on benchmark datasets.
分子动力学模拟是计算材料科学和化学中的一种重要工具,在过去十年中,它因机器学习而发生了变革。机器学习原子间势的这一快速进展在过去几年中产生了许多新架构。其中特别值得注意的是原子簇展开,它统一了许多围绕基于原子密度描述符的早期思想,以及神经等变原子间势(NequIP),这是一种具有等变特征的消息传递神经网络,在当时展现出了最先进的准确性。在这里,我们构建了一个统一这些模型的数学框架:原子簇展开被扩展并重塑为多层架构中的一层,而NequIP的线性化版本被理解为一个大得多的多项式模型的特定稀疏化形式。我们的框架还提供了一个实用工具,用于系统地探索这个统一设计空间中的不同选择。通过一组研究域内和域外准确性以及远离训练数据的平滑外推的实验对NequIP进行的消融研究,揭示了哪些设计选择对于实现高精度至关重要。我们称之为BOTnet(用于体序张量网络)的NequIP的一个简化得多的版本具有可解释的架构,并在基准数据集上保持其准确性。