Stofella Michele, Seetaloo Neeleema, St John Alexander N, Paci Emanuele, Phillips Jonathan J, Sobott Frank
School of Molecular and Cellular Biology and Astbury Centre, University of Leeds, Leeds LS2 9JT, U.K.
Living Systems Institute, University of Exeter, Exeter EX4 4QD, U.K.
Anal Chem. 2025 Feb 11;97(5):2648-2657. doi: 10.1021/acs.analchem.4c03631. Epub 2025 Jan 29.
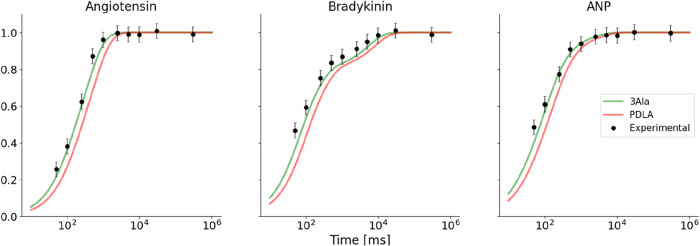
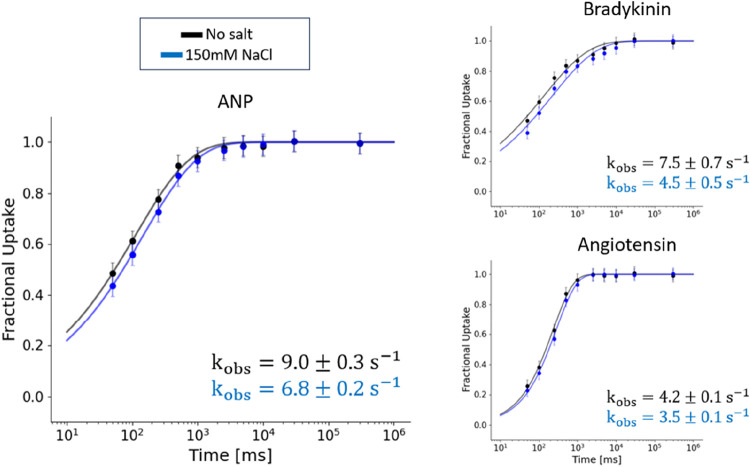
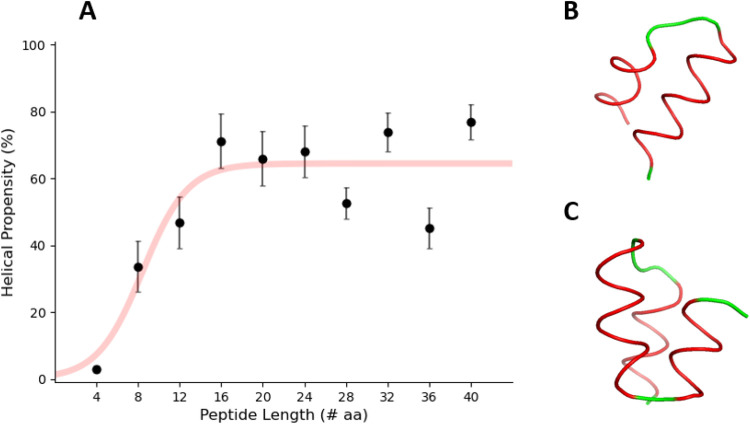
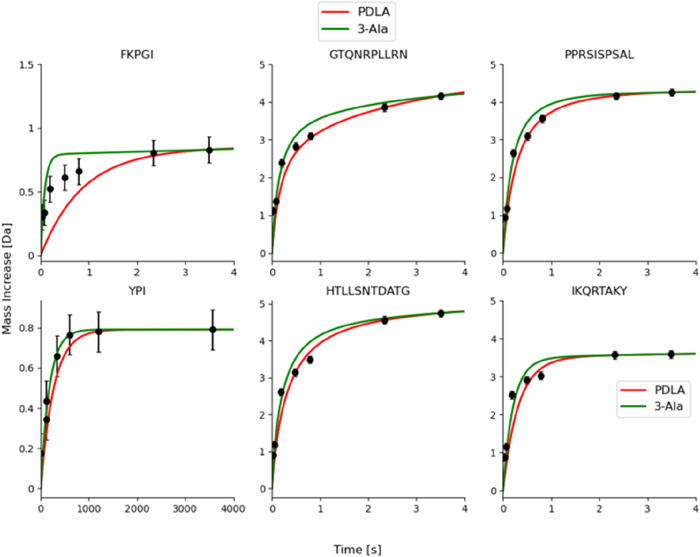
Hydrogen/deuterium exchange mass spectrometry (HDX-MS) is a powerful technique to interrogate protein structure and dynamics. With the ability to study almost any protein without a size limit, including intrinsically disordered ones, HDX-MS has shown fast growing importance as a complement to structural elucidation techniques. Current experiments compare two or more related conditions (sequences, interaction partners, excipients, conformational states, etc.) to determine statistically significant differences at a number of fixed time points and highlight areas of changed structural dynamics in the protein. The work presented here builds on the fundamental research performed in the early days of the technique and re-examines exchange rate calculations with the aim of establishing HDX-MS as an and , rather than and , measurement. We performed millisecond HDX-MS experiments on a mixture of three unstructured peptides (angiotensin, bradykinin, and atrial natriuretic peptide amide rat) and compared experimental deuterium uptake curves with theoretical ones predicted using established exchange rate calculations. With poly-dl-alanine (PDLA) commonly used as a reference, we find that experimental rates are sometimes faster than theoretically possible, while they agree much better, and are never faster, with the fully unstructured trialanine peptide (3-Ala). Molecular dynamics (MD) simulations confirm the high helical propensity of the longer and partially structured PDLA peptides, which need as few as 15 residues to form a stable helix and are therefore not suitable as an unstructured reference. Reanalysis of previously published data by Weis et al. at 100 mM NaCl however still shows a discrepancy with predictions based on 3-Ala in the absence of salt, highlighting the need for a better understanding of salt effects on exchange rates. Such currently unquantifiable salt effects prevent us from proposing a comprehensive, universal calibration framework at the moment. Nevertheless, an accurate recalibration of intrinsic exchange rate calculations is crucial to enable kinetic modeling of the exchange process and to ultimately allow HDX-MS to move toward a direct link with atomistic structural models.
氢/氘交换质谱法(HDX-MS)是一种用于研究蛋白质结构和动力学的强大技术。由于能够研究几乎任何大小不受限制的蛋白质,包括内在无序的蛋白质,HDX-MS作为结构解析技术的补充,其重要性与日俱增。当前的实验比较两个或多个相关条件(序列、相互作用伙伴、辅料、构象状态等),以在多个固定时间点确定具有统计学意义的差异,并突出蛋白质中结构动力学发生变化的区域。本文所呈现的工作基于该技术早期进行的基础研究,并重新审视了交换率计算,旨在将HDX-MS确立为一种和,而非和,测量方法。我们对三种无结构肽(血管紧张素、缓激肽和大鼠心房利钠肽酰胺)的混合物进行了毫秒级HDX-MS实验,并将实验得到的氘摄取曲线与使用既定交换率计算预测的理论曲线进行了比较。以常用作参考的聚-dl-丙氨酸(PDLA)为例,我们发现实验速率有时比理论上可能的要快,而与完全无结构的三丙氨酸肽(3-Ala)相比,它们的一致性要好得多,且从不更快。分子动力学(MD)模拟证实了较长且部分结构化的PDLA肽具有较高的螺旋倾向,这些肽只需15个残基就能形成稳定的螺旋,因此不适合作为无结构参考。然而,Weis等人在100 mM NaCl条件下对先前发表数据的重新分析仍然显示,在无盐情况下与基于3-Ala的预测存在差异,这凸显了更好地理解盐对交换率影响的必要性。目前这种无法量化的盐效应使我们目前无法提出一个全面、通用的校准框架。尽管如此,对内在交换率计算进行准确的重新校准对于实现交换过程的动力学建模以及最终使HDX-MS与原子结构模型建立直接联系至关重要。