Hakim Jill M C, Guiterrez Sneider A G, Duran Angel, Malaga-Machaca Edith, Duque Carolina, Singer Lulu, Colanzi Rony, Sherbuk Jacqueline E, Bern Caryn, Gilman Robert H, Messenger Lousia A, Mugnier Monica R
Department of Molecular Microbiology and Immunology, Johns Hopkins Bloomberg School of Public Health.
Department of International Health, Johns Hopkins Bloomberg School of Public Health.
bioRxiv. 2025 Jun 4:2025.06.04.657671. doi: 10.1101/2025.06.04.657671.
causes Chagas disease, a poorly understood and clinically heterogeneous disease. Recent work has demonstrated that parasites adapted to laboratory conditions are genomically variable, but little is known of the extent of genomic diversity from clinically isolated specimens.
In this retrospective observational genomic study, we isolated 15 specimens from three clinical studies of Chagas disease, representing different clinical contexts. We sequenced the genome of each strain and used single nucleotide variant (SNV) based analyses to estimate parasite genetic lineage, genomic population structure, regions of copy number plasticity, and to identify gene conversion events. In addition, we generated and annotated whole genome assemblies of each isolate. From these assemblies, we compared the repertoires of genes encoding for highly virulent and variable proteins that have been implicated in disease pathogenesis.
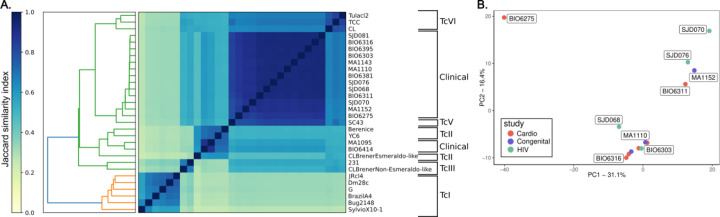
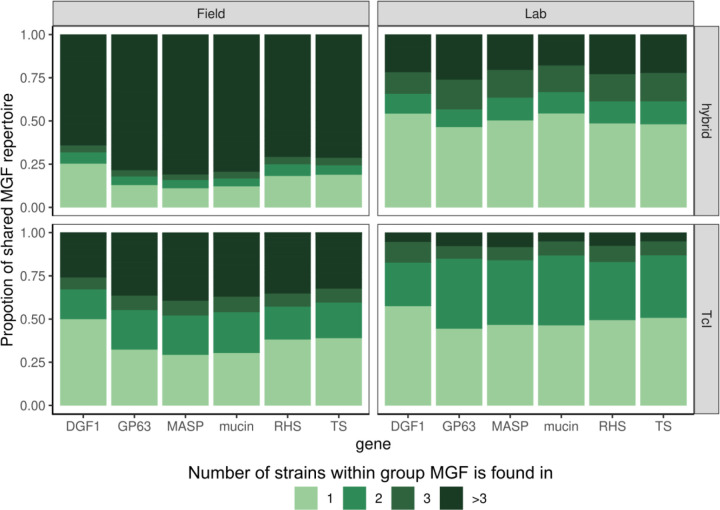
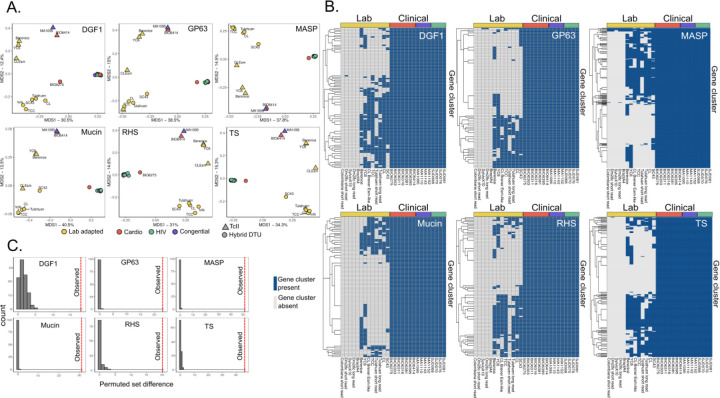
We identified parasites from two genetic lineages in this collection of clinical isolates. Our analysis revealed evidence of genomic instability. Diversity-generating copy number variation was statistically enriched in regions encoding the virulence-associated multigene families, while diversity-eliminating gene conversion events were enriched in regions depleted of multigene family members. We also discovered a set of multigene family members that is present in all of the clinically isolated parasite genomes and absent from all of the lab adapted strains, regardless of parasite lineage. Multigene family repertoires were more conserved among field isolated specimens of the same genetic lineage than among culture adapted strains of the same genetic type.
This study provides whole genome sequencing data for TcV parasites isolated from naturally infected human patients with Chagas disease for the first time. Our analysis of these genomes revealed substantial genomic instability, suggesting the parasite undergoes genomic change in response to the pressures imposed by the host environment. Moreover, we observed a set of virulence-associated genes that are present exclusively within clinical isolates and absent from lab-adapted strains, indicating a potential role for these genes in parasite survival in natural hosts. These findings highlight the limitations of genetic studies focused exclusively on lab-adapted parasite strains and provide insight into the genomic features of that are likely to be important for clinical infection.
引发恰加斯病,这是一种了解甚少且临床异质性的疾病。近期研究表明,适应实验室条件的寄生虫在基因组上具有变异性,但对于临床分离标本的基因组多样性程度知之甚少。
在这项回顾性观察基因组研究中,我们从三项恰加斯病临床研究中分离出15个标本,代表不同临床情况。我们对每个菌株的基因组进行测序,并使用基于单核苷酸变异(SNV)的分析来估计寄生虫遗传谱系、基因组群体结构、拷贝数可塑性区域,并识别基因转换事件。此外,我们生成并注释了每个分离株的全基因组组装。从这些组装中,我们比较了编码与疾病发病机制相关的高毒力和可变蛋白的基因库。
我们在这组临床分离株中鉴定出了来自两个遗传谱系的寄生虫。我们的分析揭示了基因组不稳定的证据。产生多样性的拷贝数变异在编码与毒力相关的多基因家族的区域中在统计学上更为丰富,而消除多样性的基因转换事件在多基因家族成员缺失的区域中更为丰富。我们还发现了一组多基因家族成员,它们存在于所有临床分离的寄生虫基因组中,而在所有实验室适应菌株中均不存在,无论寄生虫谱系如何。与相同遗传类型的培养适应菌株相比,相同遗传谱系的野外分离标本中的多基因家族库更为保守。
本研究首次为从自然感染恰加斯病的人类患者中分离出的TcV寄生虫提供了全基因组测序数据。我们对这些基因组的分析揭示了大量的基因组不稳定性,表明寄生虫会因宿主环境施加的压力而发生基因组变化。此外,我们观察到一组仅存在于临床分离株中而在实验室适应菌株中不存在的与毒力相关的基因,表明这些基因在寄生虫在自然宿主中的生存中可能发挥作用。这些发现凸显了仅专注于实验室适应寄生虫菌株的遗传研究的局限性,并为可能对临床感染很重要的基因组特征提供了见解。