Yap Yee Leng, Zhang Xue Wu, Danchin Antoine
HKU-Pasteur Research Centre, Dexter H,C, Man Building, 8 Sassoon Road Pokfulam, Hong Kong.
BMC Bioinformatics. 2003 Sep 20;4:43. doi: 10.1186/1471-2105-4-43.
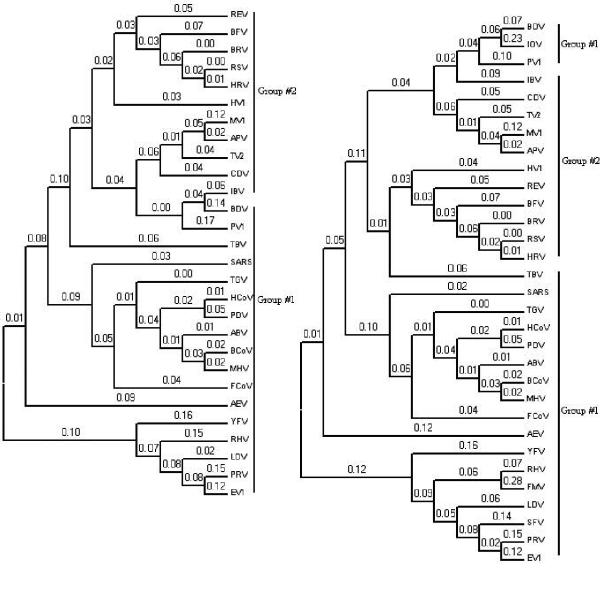
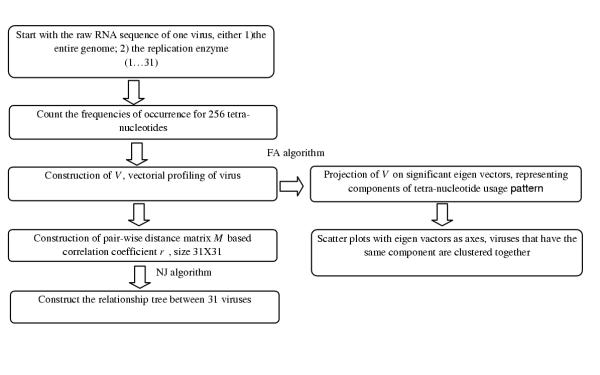
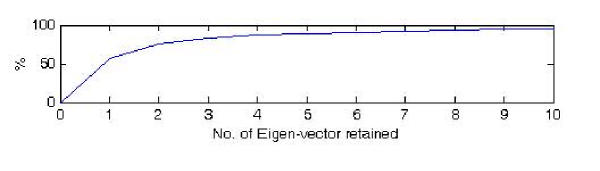
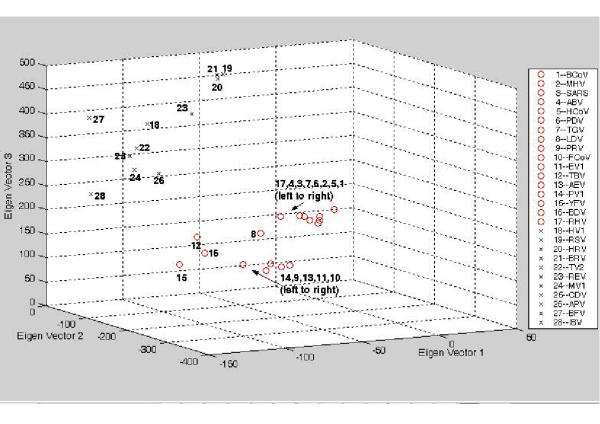
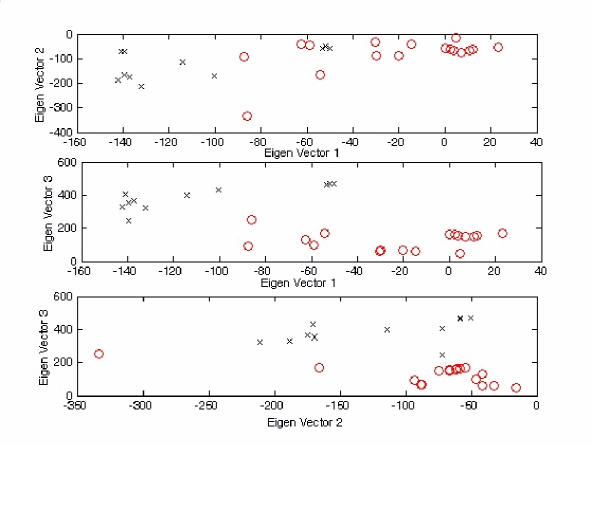
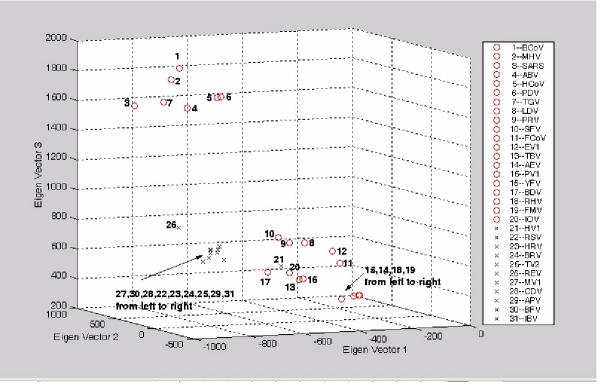
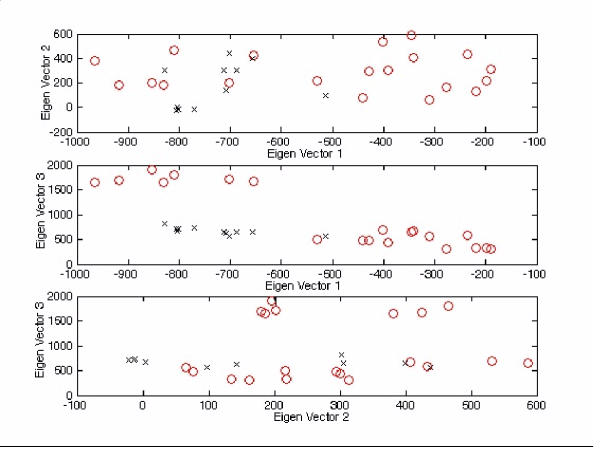
The exact origin of the cause of the Severe Acute Respiratory Syndrome (SARS) is still an open question. The genomic sequence relationship of SARS-CoV with 30 different single-stranded RNA (ssRNA) viruses of various families was studied using two non-standard approaches. Both approaches began with the vectorial profiling of the tetra-nucleotide usage pattern V for each virus. In approach one, a distance measure of a vector V, based on correlation coefficient was devised to construct a relationship tree by the neighbor-joining algorithm. In approach two, a multivariate factor analysis was performed to derive the embedded tetra-nucleotide usage patterns. These patterns were subsequently used to classify the selected viruses.
Both approaches yielded relationship outcomes that are consistent with the known virus classification. They also indicated that the genome of RNA viruses from the same family conform to a specific pattern of word usage. Based on the correlation of the overall tetra-nucleotide usage patterns, the Transmissible Gastroenteritis Virus (TGV) and the Feline CoronaVirus (FCoV) are closest to SARS-CoV. Surprisingly also, the RNA viruses that do not go through a DNA stage displayed a remarkable discrimination against the CpG and UpA di-nucleotide (z = -77.31, -52.48 respectively) and selection for UpG and CpA (z = 65.79,49.99 respectively). Potential factors influencing these biases are discussed.
The study of genomic word usage is a powerful method to classify RNA viruses. The congruence of the relationship outcomes with the known classification indicates that there exist phylogenetic signals in the tetra-nucleotide usage patterns, that is most prominent in the replicase open reading frames.
严重急性呼吸综合征(SARS)的确切病因仍未明确。采用两种非标准方法研究了严重急性呼吸综合征冠状病毒(SARS-CoV)与30种不同单链RNA(ssRNA)病毒的基因组序列关系。两种方法均从对每种病毒的四核苷酸使用模式V进行向量分析开始。在方法一中,设计了一种基于相关系数的向量V距离度量,通过邻接法构建关系树。在方法二中,进行多因素分析以推导嵌入的四核苷酸使用模式。随后,这些模式被用于对选定的病毒进行分类。
两种方法得出的关系结果均与已知的病毒分类一致。它们还表明,同一家族的RNA病毒基因组符合特定的词使用模式。基于整体四核苷酸使用模式的相关性,猪传染性胃肠炎病毒(TGV)和猫冠状病毒(FCoV)与SARS-CoV最为接近。同样令人惊讶的是,不经过DNA阶段的RNA病毒对CpG和UpA二核苷酸表现出明显的排斥(z分别为-77.31和-52.48),而对UpG和CpA有选择倾向(z分别为65.79和49.99)。文中讨论了影响这些偏差的潜在因素。
基因组词使用研究是一种对RNA病毒进行分类的有效方法。关系结果与已知分类的一致性表明,四核苷酸使用模式中存在系统发育信号,这在复制酶开放阅读框中最为突出。