Jeffery Ian B, Higgins Desmond G, Culhane Aedín C
Bioinformatics, Conway Institute, University College Dublin, Belfield, Dublin 4, Ireland.
BMC Bioinformatics. 2006 Jul 26;7:359. doi: 10.1186/1471-2105-7-359.
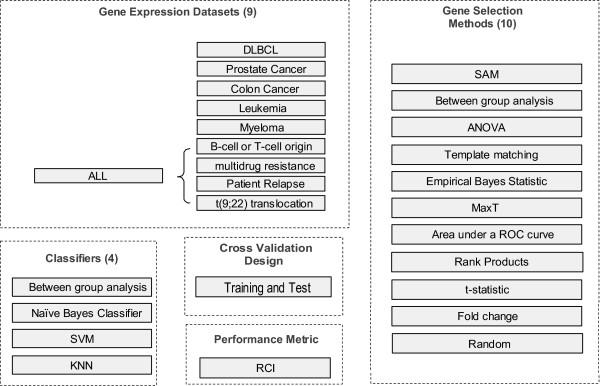
Numerous feature selection methods have been applied to the identification of differentially expressed genes in microarray data. These include simple fold change, classical t-statistic and moderated t-statistics. Even though these methods return gene lists that are often dissimilar, few direct comparisons of these exist. We present an empirical study in which we compare some of the most commonly used feature selection methods. We apply these to 9 publicly available datasets, and compare, both the gene lists produced and how these perform in class prediction of test datasets.
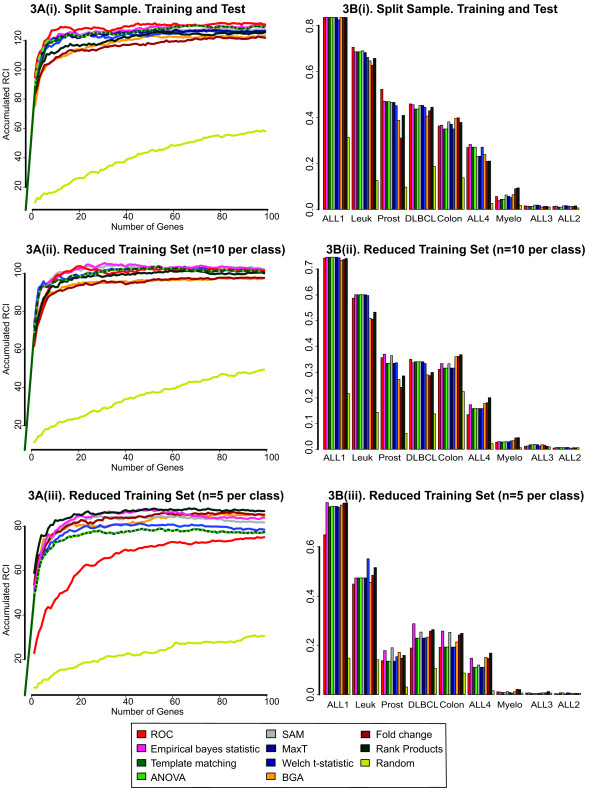
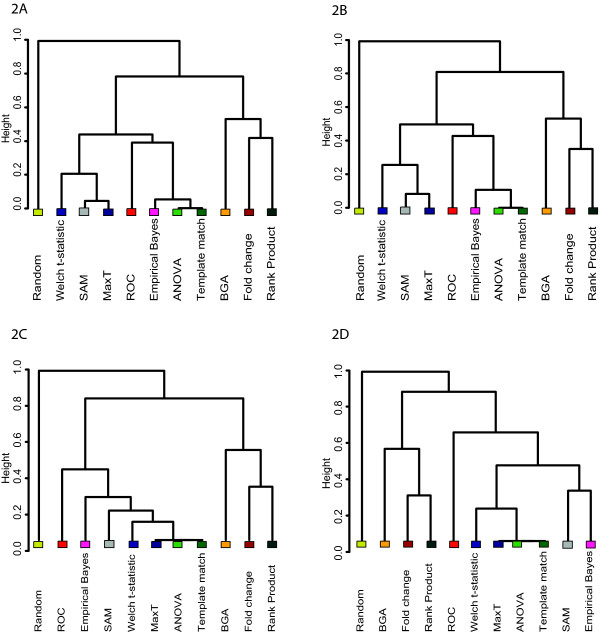
In this study, we compared the efficiency of the feature selection methods; significance analysis of microarrays (SAM), analysis of variance (ANOVA), empirical bayes t-statistic, template matching, maxT, between group analysis (BGA), Area under the receiver operating characteristic (ROC) curve, the Welch t-statistic, fold change, rank products, and sets of randomly selected genes. In each case these methods were applied to 9 different binary (two class) microarray datasets. Firstly we found little agreement in gene lists produced by the different methods. Only 8 to 21% of genes were in common across all 10 feature selection methods. Secondly, we evaluated the class prediction efficiency of each gene list in training and test cross-validation using four supervised classifiers.
We report that the choice of feature selection method, the number of genes in the genelist, the number of cases (samples) and the noise in the dataset, substantially influence classification success. Recommendations are made for choice of feature selection. Area under a ROC curve performed well with datasets that had low levels of noise and large sample size. Rank products performs well when datasets had low numbers of samples or high levels of noise. The Empirical bayes t-statistic performed well across a range of sample sizes.
众多特征选择方法已应用于识别微阵列数据中差异表达的基因。这些方法包括简单的倍数变化、经典t检验统计量和经验贝叶斯t检验统计量。尽管这些方法返回的基因列表通常不同,但很少有对它们的直接比较。我们进行了一项实证研究,比较了一些最常用的特征选择方法。我们将这些方法应用于9个公开可用的数据集,并比较生成的基因列表以及它们在测试数据集的类别预测中的表现。
在本研究中,我们比较了特征选择方法的效率;微阵列显著性分析(SAM)、方差分析(ANOVA)、经验贝叶斯t检验统计量、模板匹配、最大T值、组间分析(BGA)、受试者操作特征(ROC)曲线下面积、韦尔奇t检验统计量、倍数变化、秩乘积以及随机选择的基因集。在每种情况下,这些方法都应用于9个不同的二元(两类)微阵列数据集。首先,我们发现不同方法生成的基因列表之间几乎没有一致性。在所有10种特征选择方法中,只有8%至21%的基因是相同的。其次,我们使用四个监督分类器评估了每个基因列表在训练和测试交叉验证中的类别预测效率。
我们报告说,特征选择方法的选择、基因列表中的基因数量、样本数量以及数据集中的噪声,对分类成功有重大影响。针对特征选择的选择提出了建议。ROC曲线下面积在噪声水平低且样本量大的数据集上表现良好。当数据集样本数量少或噪声水平高时,秩乘积表现良好。经验贝叶斯t检验统计量在一系列样本量下表现良好。