Yeang Chen-Hsiang, Haussler David
Simons Center for Systems Biology, Institute for Advanced Study, Princeton, New Jersey, United States of America.
PLoS Comput Biol. 2007 Nov;3(11):e211. doi: 10.1371/journal.pcbi.0030211. Epub 2007 Sep 18.
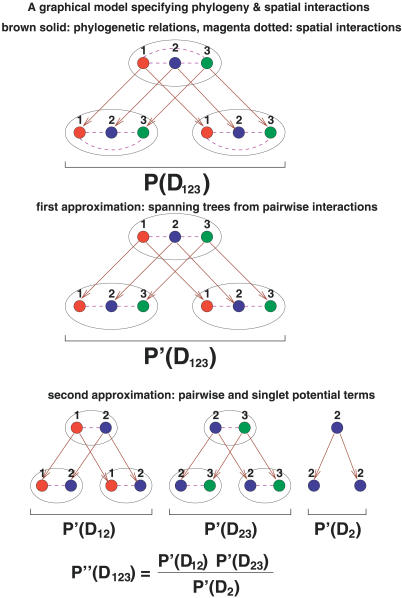
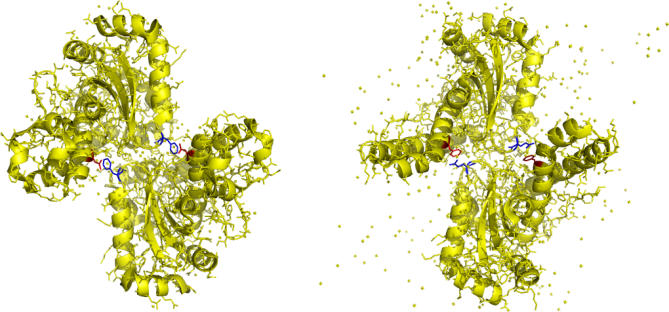
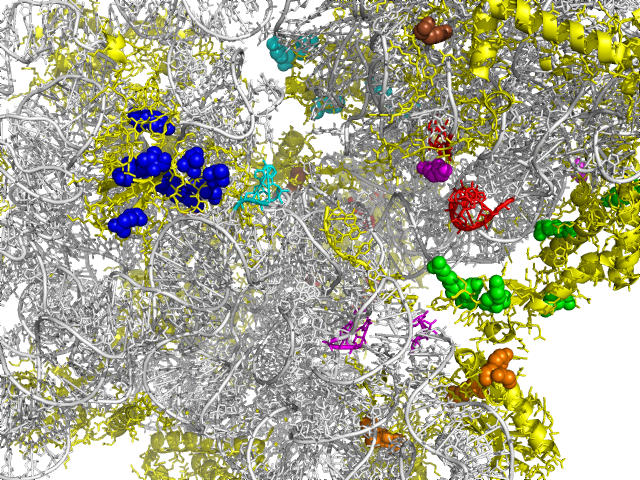
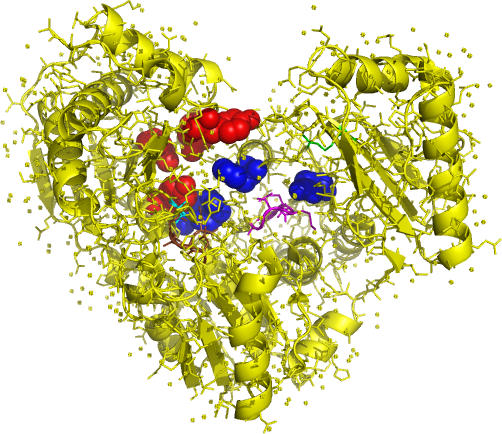
Correlated changes of nucleic or amino acids have provided strong information about the structures and interactions of molecules. Despite the rich literature in coevolutionary sequence analysis, previous methods often have to trade off between generality, simplicity, phylogenetic information, and specific knowledge about interactions. Furthermore, despite the evidence of coevolution in selected protein families, a comprehensive screening of coevolution among all protein domains is still lacking. We propose an augmented continuous-time Markov process model for sequence coevolution. The model can handle different types of interactions, incorporate phylogenetic information and sequence substitution, has only one extra free parameter, and requires no knowledge about interaction rules. We employ this model to large-scale screenings on the entire protein domain database (Pfam). Strikingly, with 0.1 trillion tests executed, the majority of the inferred coevolving protein domains are functionally related, and the coevolving amino acid residues are spatially coupled. Moreover, many of the coevolving positions are located at functionally important sites of proteins/protein complexes, such as the subunit linkers of superoxide dismutase, the tRNA binding sites of ribosomes, the DNA binding region of RNA polymerase, and the active and ligand binding sites of various enzymes. The results suggest sequence coevolution manifests structural and functional constraints of proteins. The intricate relations between sequence coevolution and various selective constraints are worth pursuing at a deeper level.
核酸或氨基酸的相关变化为分子的结构和相互作用提供了有力信息。尽管在共进化序列分析方面有丰富的文献,但先前的方法往往不得不在通用性、简单性、系统发育信息以及关于相互作用的特定知识之间进行权衡。此外,尽管在选定的蛋白质家族中有共进化的证据,但仍缺乏对所有蛋白质结构域之间共进化的全面筛选。我们提出了一种用于序列共进化的增强型连续时间马尔可夫过程模型。该模型可以处理不同类型的相互作用,纳入系统发育信息和序列替换,只有一个额外的自由参数,并且不需要关于相互作用规则的知识。我们将此模型应用于对整个蛋白质结构域数据库(Pfam)的大规模筛选。令人惊讶的是,在执行了1万亿次测试的情况下,大多数推断出的共进化蛋白质结构域在功能上是相关的,并且共进化的氨基酸残基在空间上是耦合的。此外,许多共进化位点位于蛋白质/蛋白质复合物的功能重要位点上,例如超氧化物歧化酶的亚基连接区、核糖体的tRNA结合位点、RNA聚合酶的DNA结合区域以及各种酶的活性和配体结合位点。结果表明序列共进化体现了蛋白质的结构和功能限制。序列共进化与各种选择限制之间的复杂关系值得在更深层次上进行探究。