Moukhametzianov Rouslan, Burghammer Manfred, Edwards Patricia C, Petitdemange Sebastien, Popov Dimitri, Fransen Maikel, McMullan Gregory, Schertler Gebhard F X, Riekel Christian
MRC Laboratory of Molecular Biology, Hills Road, Cambridge CB2 0QH, England.
Acta Crystallogr D Biol Crystallogr. 2008 Feb;64(Pt 2):158-66. doi: 10.1107/S090744490705812X. Epub 2008 Jan 16.
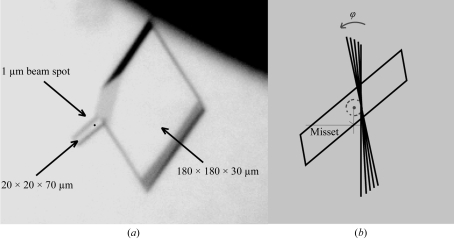
For the first time, protein microcrystallography has been performed with a focused synchrotron-radiation beam of 1 microm using a goniometer with a sub-micrometre sphere of confusion. The crystal structure of xylanase II has been determined with a flux density of about 3 x 10(10) photons s(-1) microm(-2) at the sample. Two sets of diffraction images collected from different sized crystals were shown to comprise data of good quality, which allowed a 1.5 A resolution xylanase II structure to be obtained. The main conclusion of this experiment is that a high-resolution diffraction pattern can be obtained from 20 microm(3) crystal volume, corresponding to about 2 x 10(8) unit cells. Despite the high irradiation dose in this case, it was possible to obtain an excellent high-resolution map and it could be concluded from the individual atomic B-factor patterns that there was no evidence of significant radiation damage. The photoelectron escape from a narrow diffraction channel is a possible reason for reduced radiation damage as indicated by Monte Carlo simulations. These results open many new opportunities in scanning protein microcrystallography and make random data collection from microcrystals a real possibility, therefore enabling structures to be solved from much smaller crystals than previously anticipated as long as the crystallites are well ordered.
首次使用带有亚微米级模糊球的测角仪,以1微米的聚焦同步辐射光束进行了蛋白质微晶学研究。在样品处通量密度约为3×10¹⁰光子·秒⁻¹·微米⁻²的条件下,确定了木聚糖酶II的晶体结构。从不同尺寸晶体收集的两组衍射图像显示包含高质量数据,从而获得了分辨率为1.5埃的木聚糖酶II结构。该实验的主要结论是,可以从20微米³的晶体体积(对应约2×10⁸个晶胞)获得高分辨率衍射图样。尽管在这种情况下辐照剂量很高,但仍有可能获得出色的高分辨率图谱,并且从各个原子的B因子图样可以得出结论,没有明显辐射损伤的证据。蒙特卡罗模拟表明,光电子从狭窄衍射通道逸出可能是辐射损伤降低的一个原因。这些结果为扫描蛋白质微晶学开辟了许多新机会,并使从微晶中随机收集数据成为现实,因此只要微晶排列良好,就能够从比以前预期小得多的晶体中解析出结构。