Cho Young-Rae, Shi Lei, Ramanathan Murali, Zhang Aidong
Department of Computer Science and Engineering, State University of New York, Buffalo, NY, USA.
BMC Bioinformatics. 2008 Sep 18;9:382. doi: 10.1186/1471-2105-9-382.
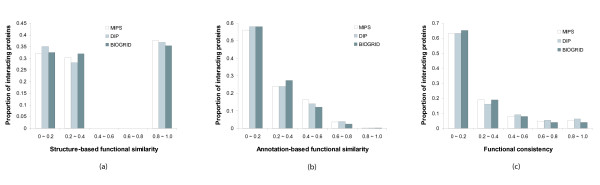
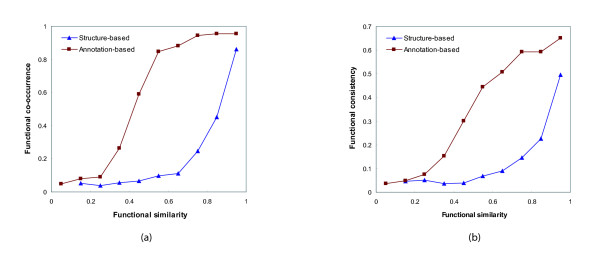
The functional characterization of newly discovered proteins has been a challenge in the post-genomic era. Protein-protein interactions provide insights into the functional analysis because the function of unknown proteins can be postulated on the basis of their interaction evidence with known proteins. The protein-protein interaction data sets have been enriched by high-throughput experimental methods. However, the functional analysis using the interaction data has a limitation in accuracy because of the presence of the false positive data experimentally generated and the interactions that are a lack of functional linkage.
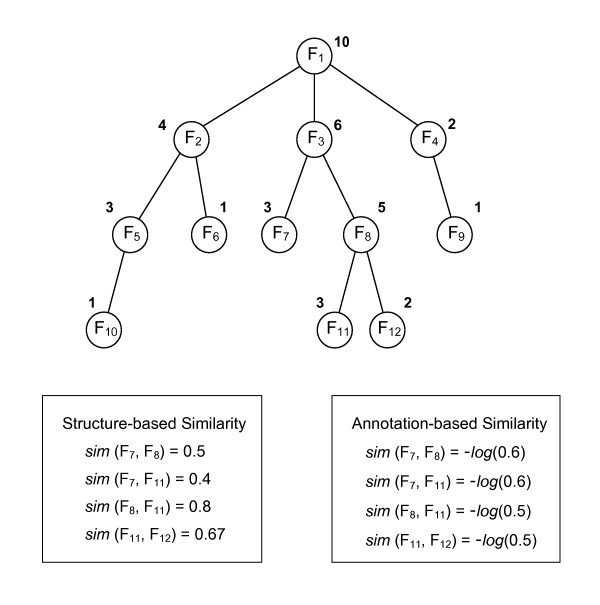
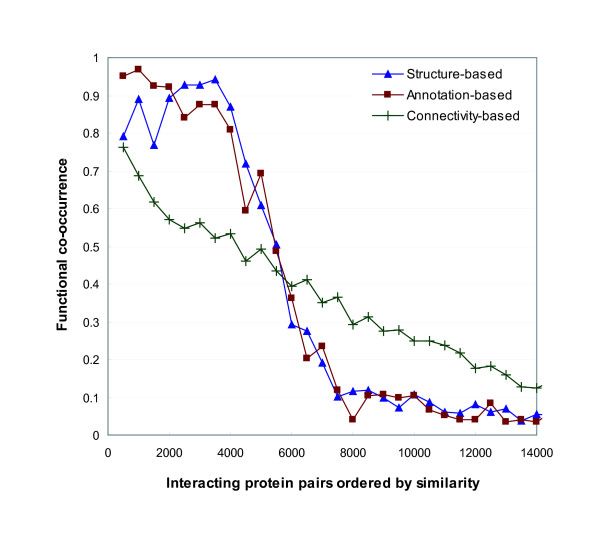
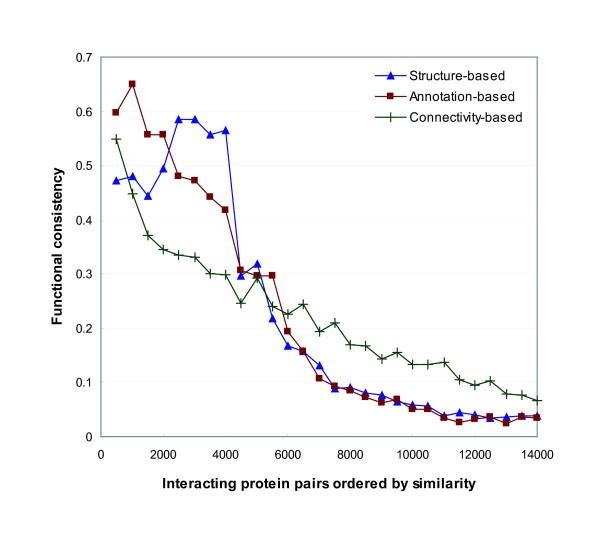
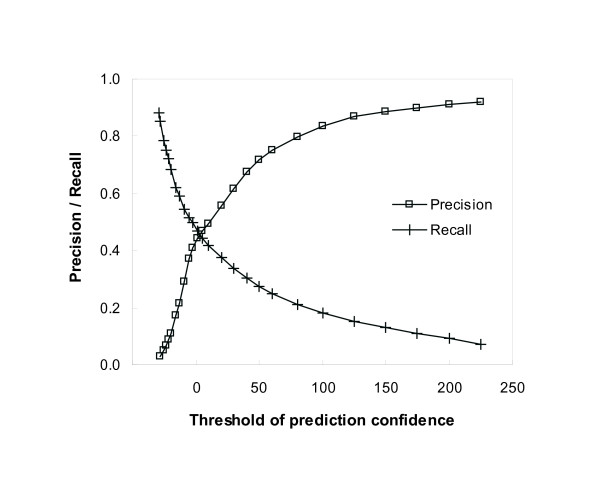
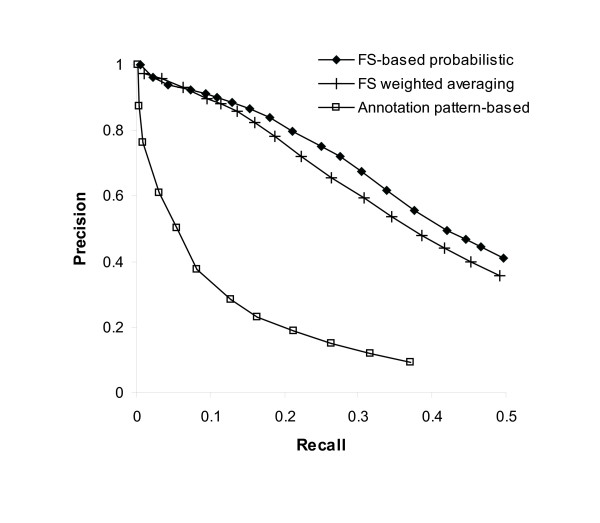
Protein-protein interaction data can be integrated with the functional knowledge existing in the Gene Ontology (GO) database. We apply similarity measures to assess the functional similarity between interacting proteins. We present a probabilistic framework for predicting functions of unknown proteins based on the functional similarity. We use the leave-one-out cross validation to compare the performance. The experimental results demonstrate that our algorithm performs better than other competing methods in terms of prediction accuracy. In particular, it handles the high false positive rates of current interaction data well.
The experimentally determined protein-protein interactions are erroneous to uncover the functional associations among proteins. The performance of function prediction for uncharacterized proteins can be enhanced by the integration of multiple data sources available.
在后基因组时代,新发现蛋白质的功能表征一直是一项挑战。蛋白质 - 蛋白质相互作用为功能分析提供了见解,因为未知蛋白质的功能可以根据它们与已知蛋白质的相互作用证据来推测。蛋白质 - 蛋白质相互作用数据集已通过高通量实验方法得到丰富。然而,由于实验产生的假阳性数据以及缺乏功能联系的相互作用的存在,使用相互作用数据进行功能分析在准确性方面存在局限性。
蛋白质 - 蛋白质相互作用数据可以与基因本体论(GO)数据库中存在的功能知识相结合。我们应用相似性度量来评估相互作用蛋白质之间的功能相似性。我们提出了一个基于功能相似性预测未知蛋白质功能的概率框架。我们使用留一法交叉验证来比较性能。实验结果表明,我们的算法在预测准确性方面比其他竞争方法表现更好。特别是,它能很好地处理当前相互作用数据的高假阳性率。
实验确定的蛋白质 - 蛋白质相互作用对于揭示蛋白质之间的功能关联是有误差的。通过整合可用的多个数据源,可以提高对未表征蛋白质的功能预测性能。