Department of Neurology, Yale University School of Medicine, New Haven, CT 06510, USA.
Mol Pain. 2010 Jun 8;6:35. doi: 10.1186/1744-8069-6-35.
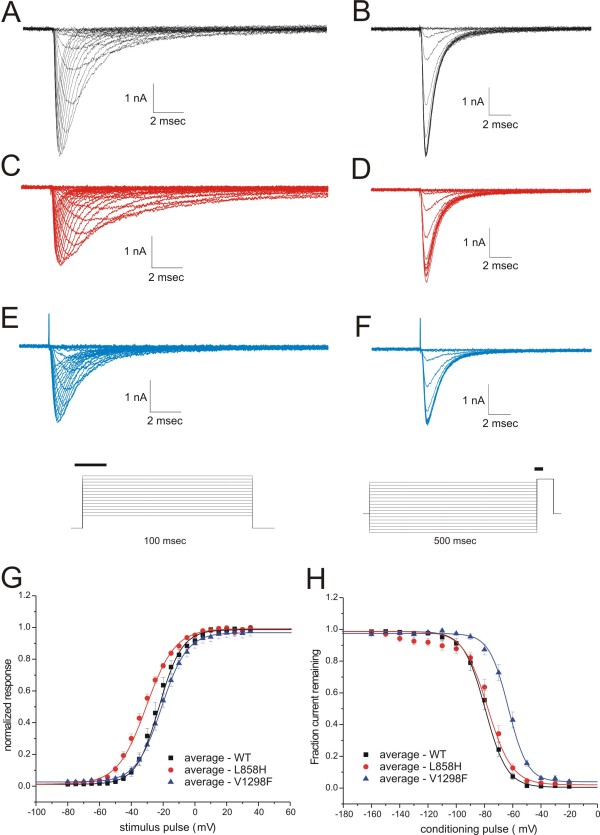
A direct role of sodium channels in pain has recently been confirmed by establishing a monogenic link between SCN9A, the gene which encodes sodium channel Nav1.7, and pain disorders in humans, with gain-of-function mutations causing severe pain syndromes, and loss-of-function mutations causing congenital indifference to pain. Expression of sodium channel Nav1.8 in DRG neurons has also been shown to be essential for the manifestation of mutant Nav1.7-induced neuronal hyperexcitability. These findings have confirmed key roles of Nav1.7 and Nav1.8 in pain and identify these channels as novel targets for pain therapeutic development. Ranolazine preferentially blocks cardiac late sodium currents at concentrations that do not significantly reduce peak sodium current. Ranolazine also blocks wild-type Nav1.7 and Nav1.8 channels in a use-dependent manner. However, ranolazine's effects on gain-of-function mutations of Nav1.7 and on DRG neuron excitability have not been investigated. We used voltage- and current-clamp recordings to evaluate the hypothesis that ranolazine may be effective in regulating Nav1.7-induced DRG neuron hyperexcitability.
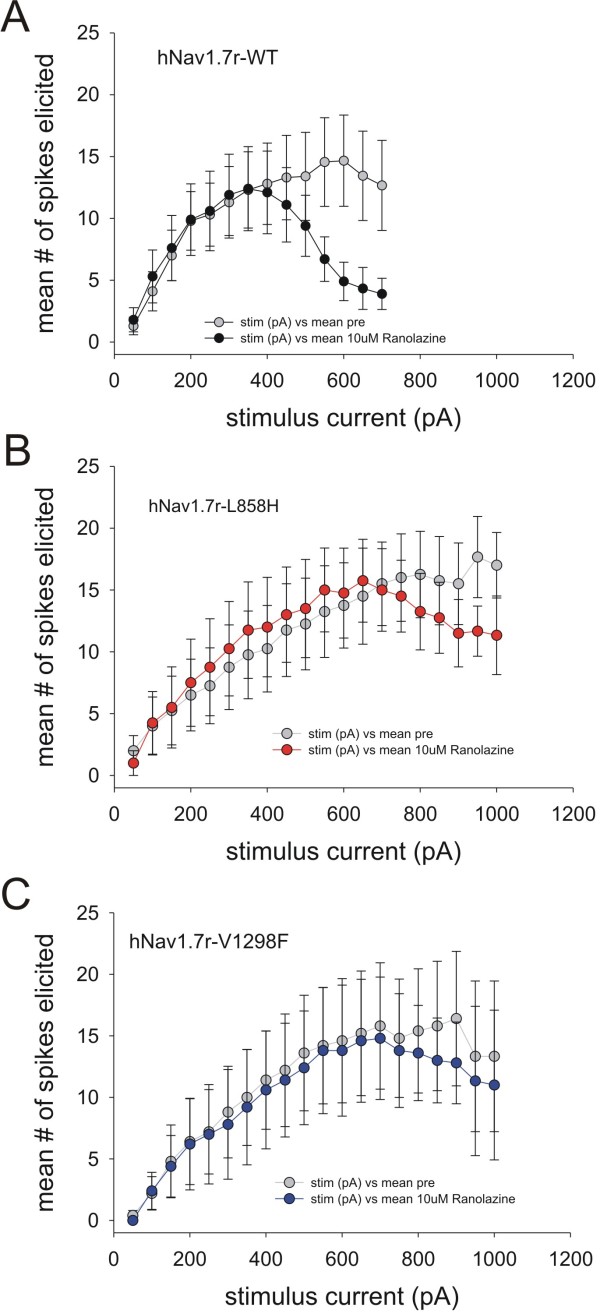
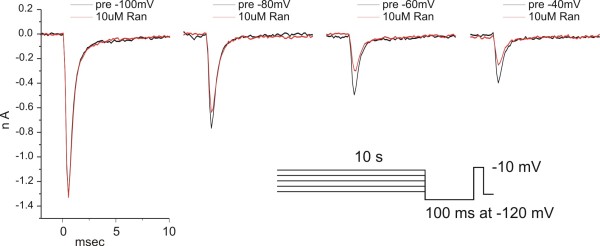
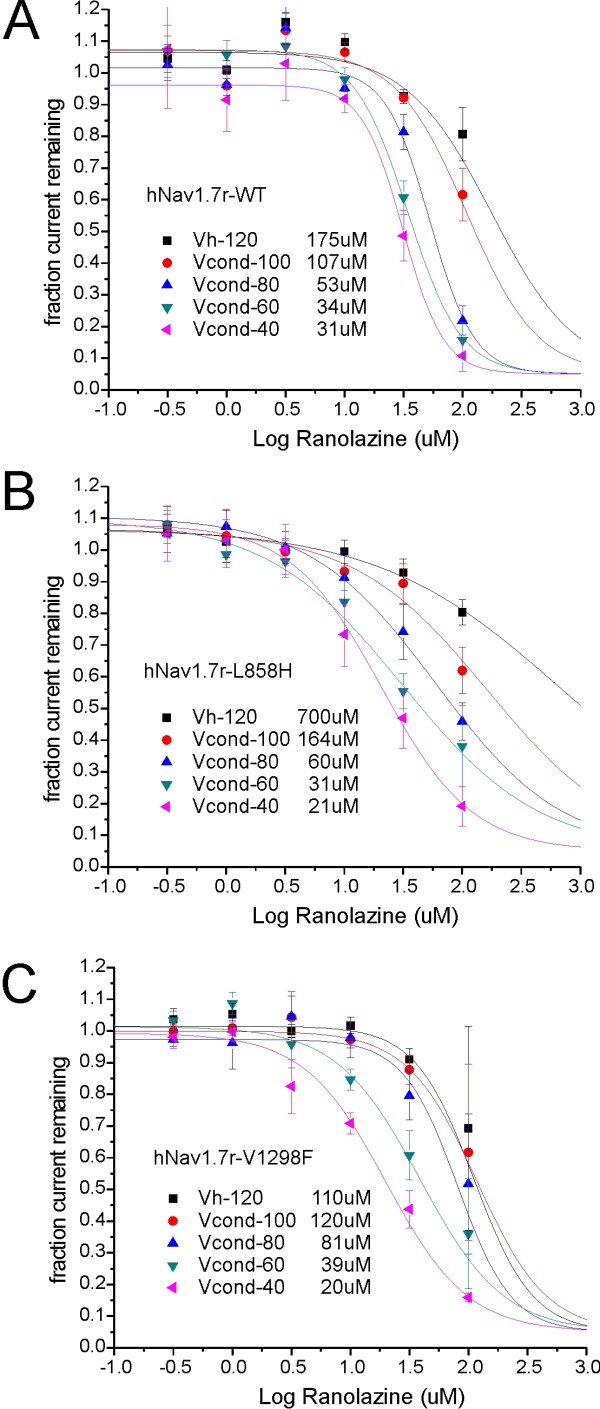
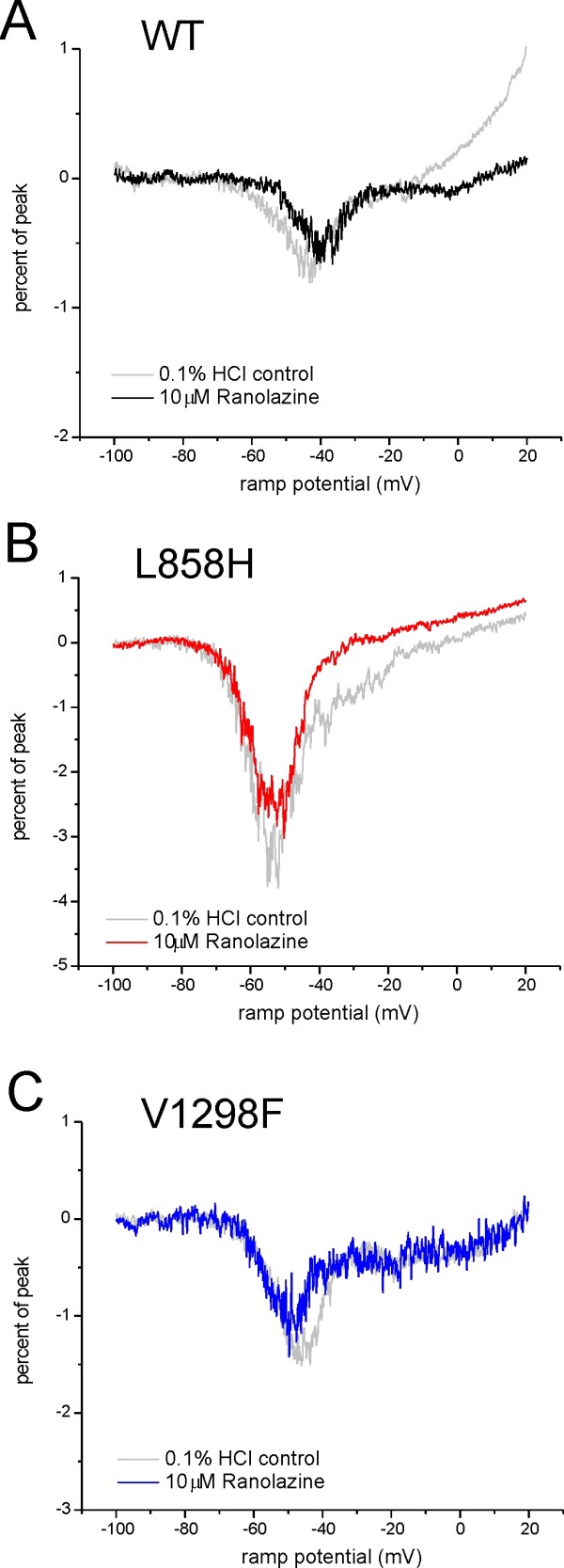
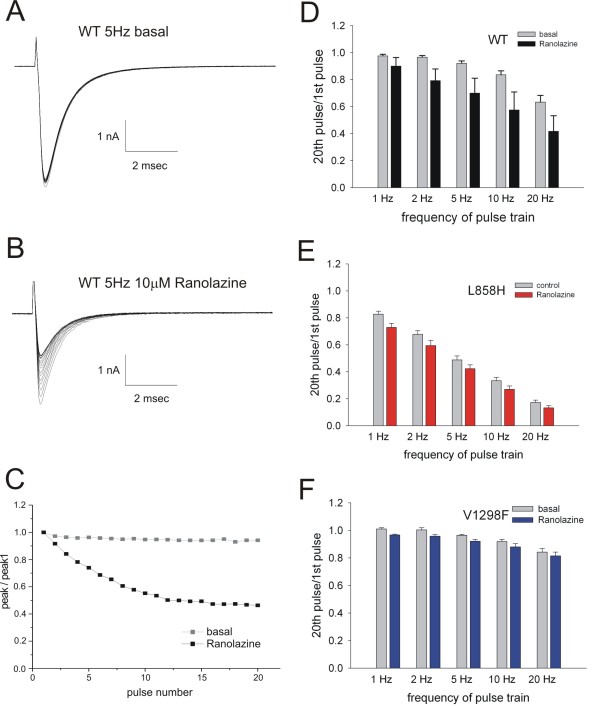
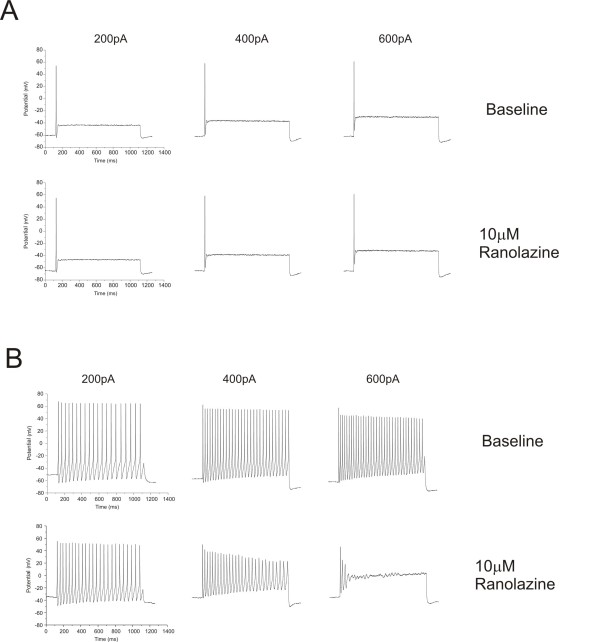
We show that ranolazine produces comparable block of peak and ramp currents of wild-type Nav1.7 and mutant Nav1.7 channels linked to Inherited Erythromelalgia and Paroxysmal Extreme Pain Disorder. We also show that ranolazine, at a clinically-relevant concentration, blocks high-frequency firing of DRG neurons expressing wild-type but not mutant channels.
Our data suggest that ranalozine can attenuate hyperexcitability of DRG neurons over-expressing wild-type Nav1.7 channels, as occurs in acquired neuropathic and inflammatory pain, and thus merits further study as an alternative to existing non-selective sodium channel blockers.
最近通过在 SCN9A(编码钠通道 Nav1.7 的基因)与人类疼痛障碍之间建立单基因联系,证实了钠通道在疼痛中的直接作用,功能获得性突变导致严重的疼痛综合征,而功能丧失性突变导致先天性无痛。DRG 神经元中钠通道 Nav1.8 的表达对于突变 Nav1.7 诱导的神经元过度兴奋的表现也至关重要。这些发现证实了 Nav1.7 和 Nav1.8 在疼痛中的关键作用,并将这些通道确定为新型疼痛治疗开发的靶点。雷诺嗪在不显著减少峰值钠电流的浓度下优先阻断心脏晚期钠电流。雷诺嗪还以使用依赖性方式阻断野生型 Nav1.7 和 Nav1.8 通道。然而,雷诺嗪对 Nav1.7 的功能获得性突变和 DRG 神经元兴奋性的影响尚未得到研究。我们使用电压和电流钳记录来评估雷诺嗪可能有效调节 Nav1.7 诱导的 DRG 神经元过度兴奋的假说。
我们表明,雷诺嗪对与遗传性红细胞增多症和阵发性剧痛障碍相关的野生型 Nav1.7 和突变型 Nav1.7 通道的峰值和斜坡电流产生可比的阻断作用。我们还表明,在临床相关浓度下,雷诺嗪阻断表达野生型而非突变型通道的 DRG 神经元的高频放电。
我们的数据表明,雷诺嗪可以减轻过度兴奋的 DRG 神经元,这些神经元过度表达野生型 Nav1.7 通道,如在获得性神经病理性和炎症性疼痛中发生的那样,因此值得进一步研究作为现有非选择性钠通道阻滞剂的替代物。