Department of Mathematics, University of California, Berkeley, California, USA.
BMC Bioinformatics. 2010 Aug 18;11:430. doi: 10.1186/1471-2105-11-430.
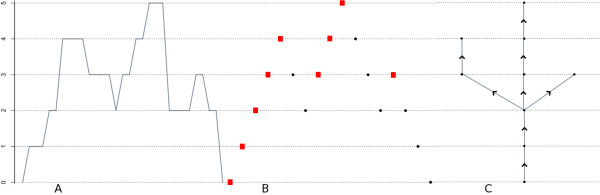
We study the statistical properties of fragment coverage in genome sequencing experiments. In an extension of the classic Lander-Waterman model, we consider the effect of the length distribution of fragments. We also introduce a coding of the shape of the coverage depth function as a tree and explain how this can be used to detect regions with anomalous coverage. This modeling perspective is especially germane to current high-throughput sequencing experiments, where both sample preparation protocols and sequencing technology particulars can affect fragment length distributions.
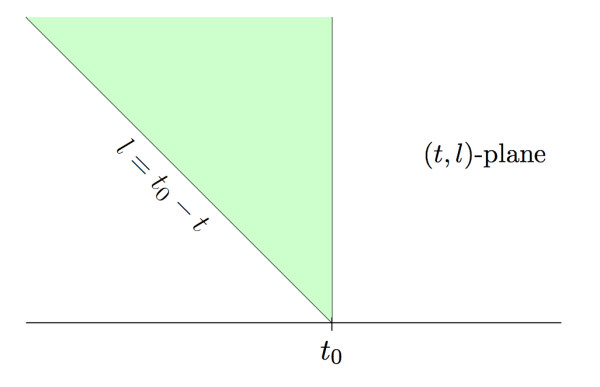
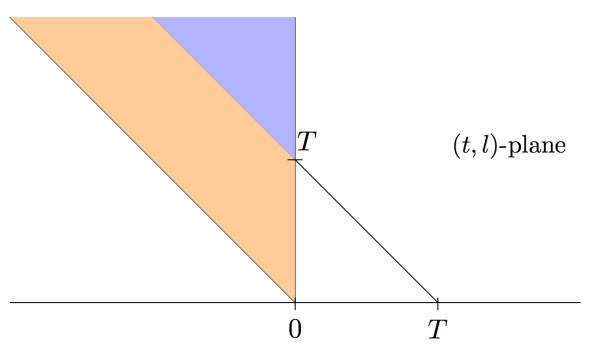
Under the mild assumptions that fragment start sites are Poisson distributed and successive fragment lengths are independent and identically distributed, we observe that, regardless of fragment length distribution, the fragments produced in a sequencing experiment can be viewed as resulting from a two-dimensional spatial Poisson process. We then study the successive jumps of the coverage function, and show that they can be encoded as a random tree that is approximately a Galton-Watson tree with generation-dependent geometric offspring distributions whose parameters can be computed.
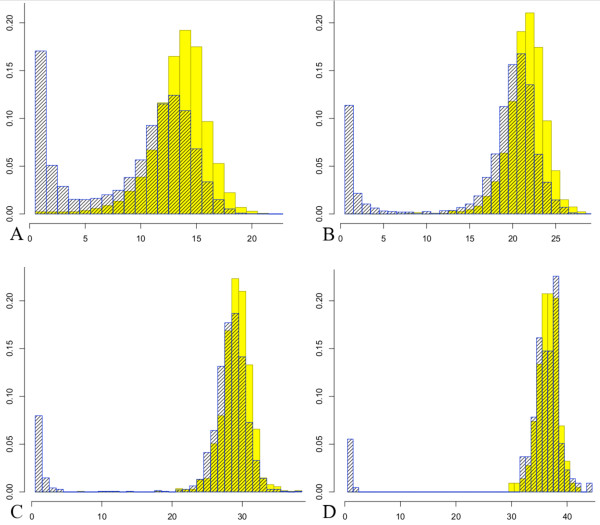
We extend standard analyses of shotgun sequencing that focus on coverage statistics at individual sites, and provide a null model for detecting deviations from random coverage in high-throughput sequence census based experiments. Our approach leads to explicit determinations of the null distributions of certain test statistics, while for others it greatly simplifies the approximation of their null distributions by simulation. Our focus on fragments also leads to a new approach to visualizing sequencing data that is of independent interest.
我们研究了基因组测序实验中片段覆盖度的统计特性。在经典的 Lander-Waterman 模型的扩展中,我们考虑了片段长度分布的影响。我们还引入了一种将覆盖深度函数形状编码为树的方法,并解释了如何使用这种方法来检测具有异常覆盖度的区域。这种建模视角尤其适用于当前的高通量测序实验,其中样品制备方案和测序技术细节都可能影响片段长度分布。
在片段起始位点呈泊松分布且连续片段长度独立同分布的温和假设下,我们观察到,无论片段长度分布如何,测序实验中产生的片段都可以看作是二维空间泊松过程的结果。然后,我们研究了覆盖函数的连续跳跃,并表明它们可以编码为随机树,该树近似于具有世代相关几何后代分布的 Galton-Watson 树,其参数可以计算。
我们扩展了专注于单个位点覆盖度统计的标准霰弹枪测序分析,并提供了一种基于高通量序列计数实验检测随机覆盖偏差的零模型。我们的方法导致了某些检验统计量的零分布的显式确定,而对于其他检验统计量,则通过模拟极大简化了它们的零分布的近似。我们对片段的关注也导致了一种新的可视化测序数据的方法,这是独立的兴趣点。