Department of Molecular Genetics, University of Toronto, Toronto, Ontario, Canada.
PLoS Pathog. 2010 Aug 26;6(8):e1001069. doi: 10.1371/journal.ppat.1001069.
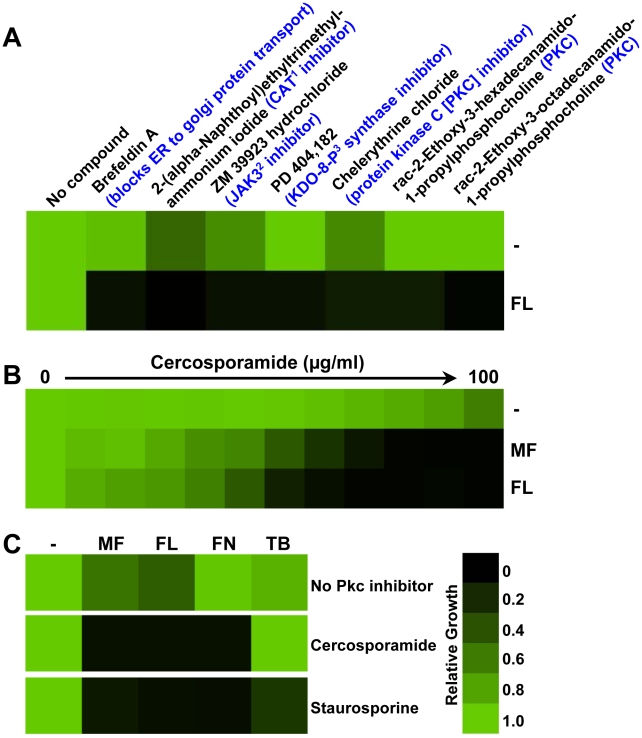
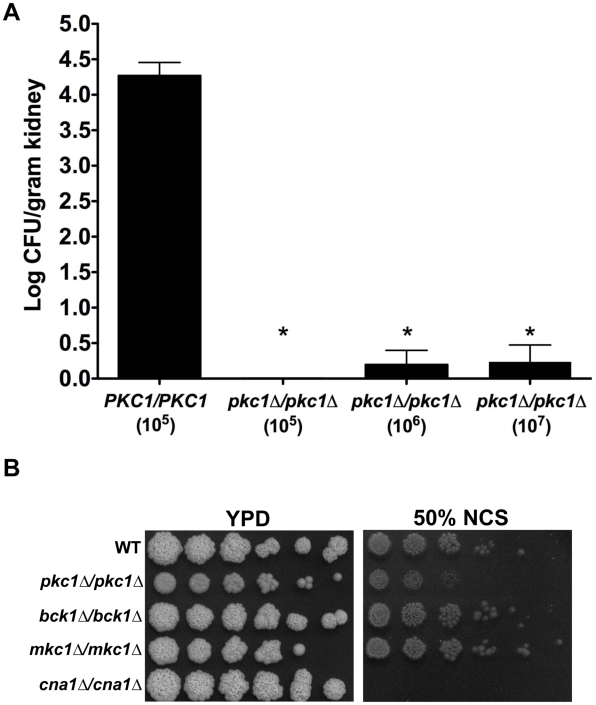
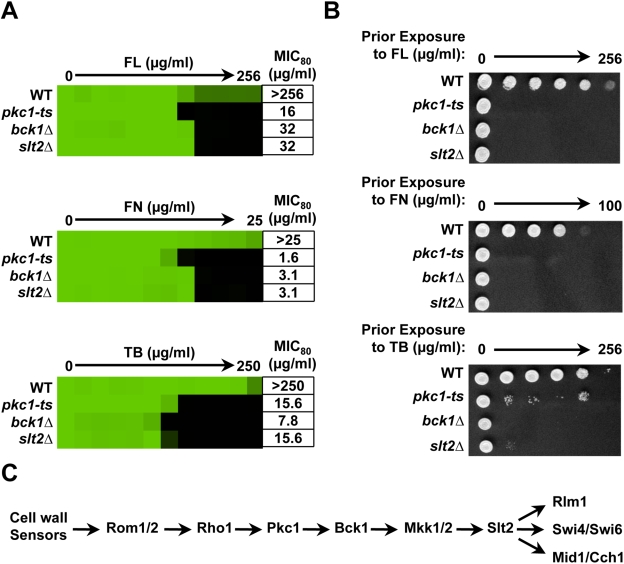
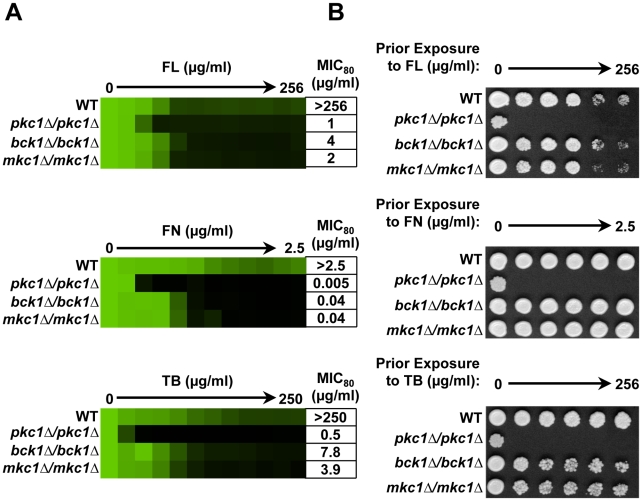
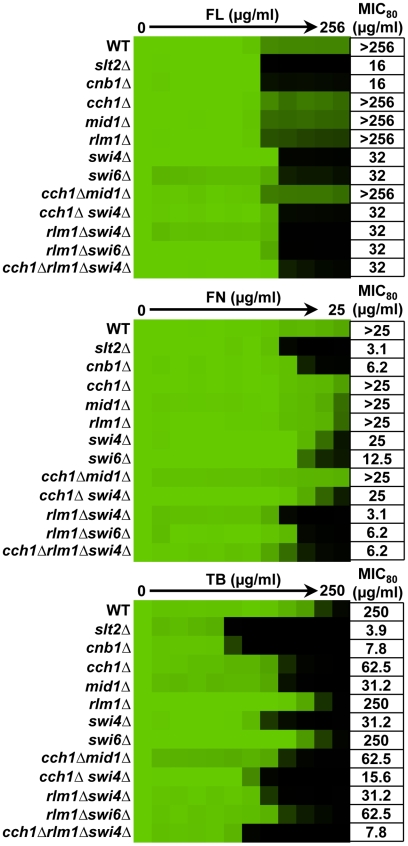
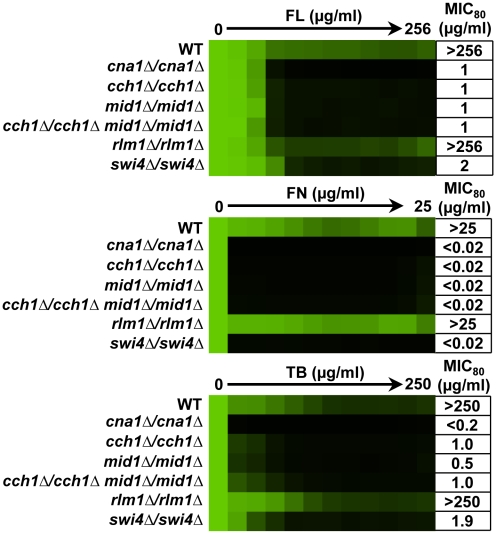
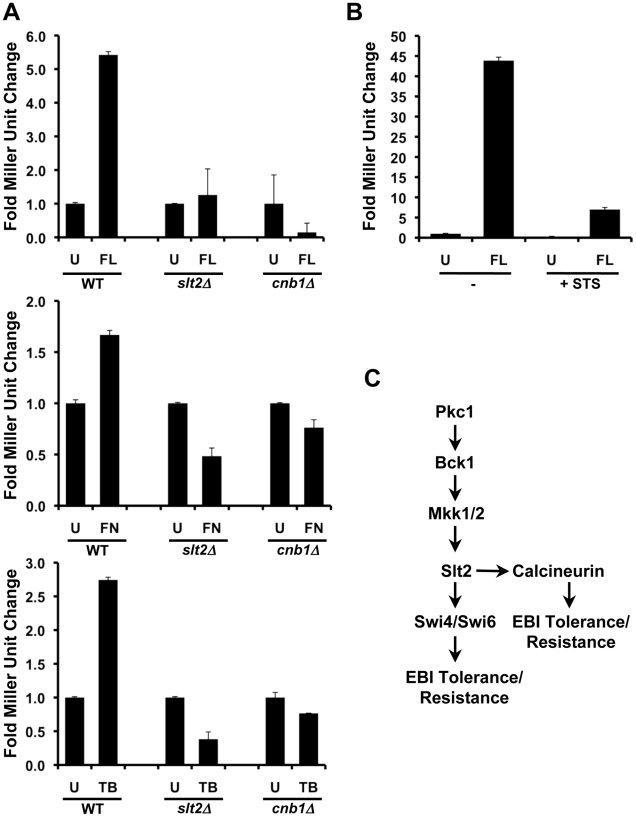
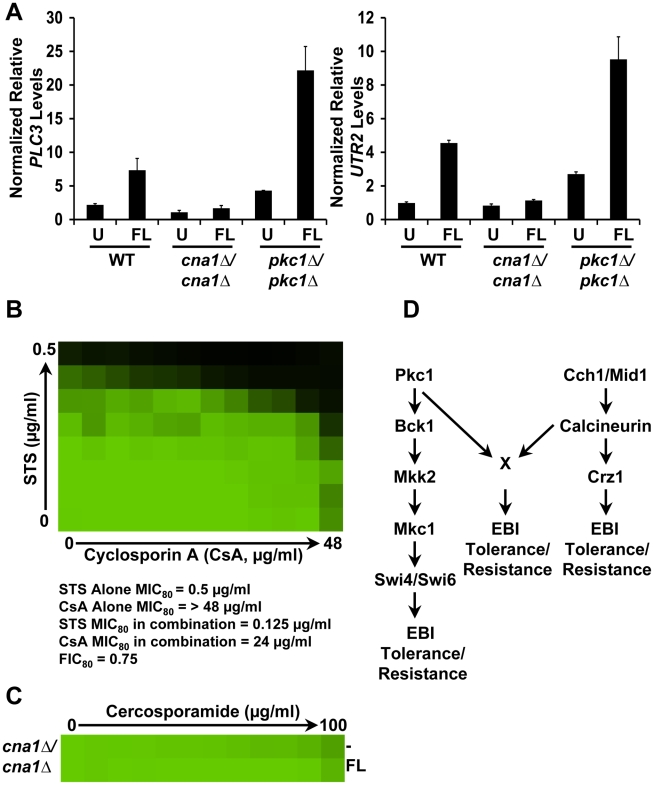
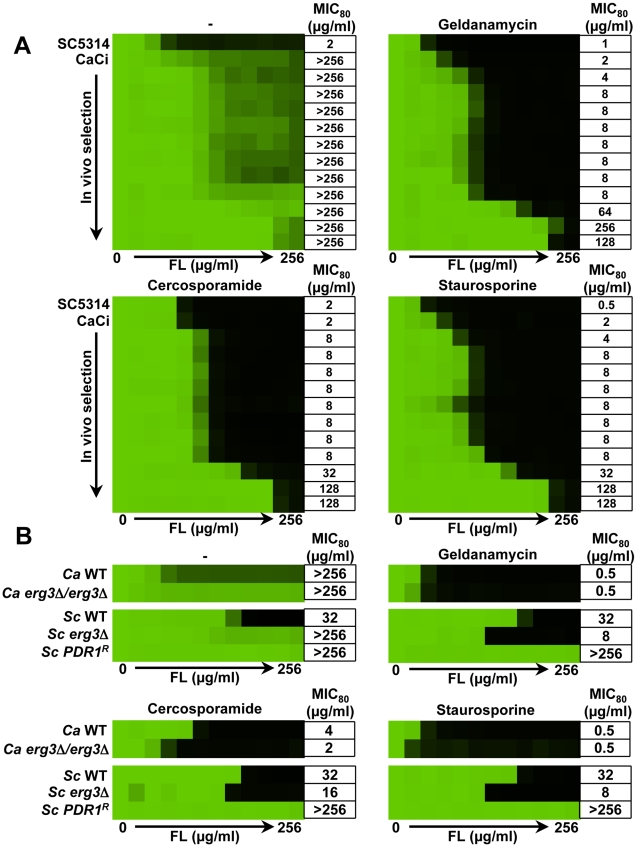
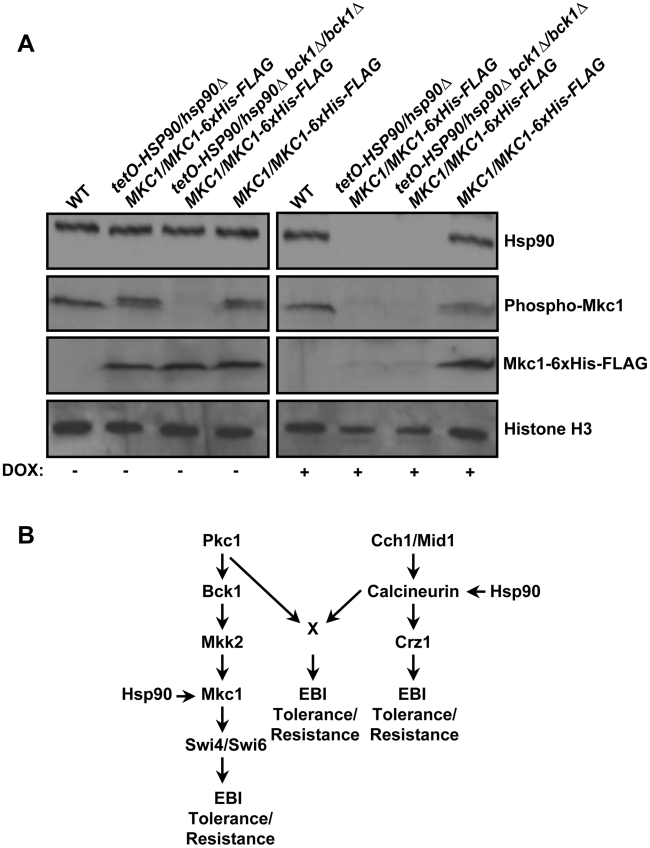
Fungal pathogens exploit diverse mechanisms to survive exposure to antifungal drugs. This poses concern given the limited number of clinically useful antifungals and the growing population of immunocompromised individuals vulnerable to life-threatening fungal infection. To identify molecules that abrogate resistance to the most widely deployed class of antifungals, the azoles, we conducted a screen of 1,280 pharmacologically active compounds. Three out of seven hits that abolished azole resistance of a resistant mutant of the model yeast Saccharomyces cerevisiae and a clinical isolate of the leading human fungal pathogen Candida albicans were inhibitors of protein kinase C (PKC), which regulates cell wall integrity during growth, morphogenesis, and response to cell wall stress. Pharmacological or genetic impairment of Pkc1 conferred hypersensitivity to multiple drugs that target synthesis of the key cell membrane sterol ergosterol, including azoles, allylamines, and morpholines. Pkc1 enabled survival of cell membrane stress at least in part via the mitogen activated protein kinase (MAPK) cascade in both species, though through distinct downstream effectors. Strikingly, inhibition of Pkc1 phenocopied inhibition of the molecular chaperone Hsp90 or its client protein calcineurin. PKC signaling was required for calcineurin activation in response to drug exposure in S. cerevisiae. In contrast, Pkc1 and calcineurin independently regulate drug resistance via a common target in C. albicans. We identified an additional level of regulatory control in the C. albicans circuitry linking PKC signaling, Hsp90, and calcineurin as genetic reduction of Hsp90 led to depletion of the terminal MAPK, Mkc1. Deletion of C. albicans PKC1 rendered fungistatic ergosterol biosynthesis inhibitors fungicidal and attenuated virulence in a murine model of systemic candidiasis. This work establishes a new role for PKC signaling in drug resistance, novel circuitry through which Hsp90 regulates drug resistance, and that targeting stress response signaling provides a promising strategy for treating life-threatening fungal infections.
真菌病原体利用多种机制来逃避抗真菌药物的作用。鉴于临床上可用的抗真菌药物数量有限,以及免疫功能低下的人群日益增加,容易受到危及生命的真菌感染,这令人担忧。为了寻找能够克服最广泛使用的一类抗真菌药物——唑类药物的耐药性的分子,我们对 1280 种具有药理活性的化合物进行了筛选。在能够消除模型酵母酿酒酵母的耐药突变体和主要人类真菌病原体白色念珠菌的临床分离株对唑类药物耐药性的 7 个命中物中,有 3 个是蛋白激酶 C(PKC)的抑制剂,PKC 在生长、形态发生和应对细胞壁应激时调节细胞壁完整性。Pkc1 的药理学或遗传学损伤赋予了对多种药物的敏感性,这些药物靶向关键细胞膜固醇麦角固醇的合成,包括唑类、烯丙胺类和吗啉类。在这两个物种中,Pkc1 至少部分通过丝裂原激活蛋白激酶(MAPK)级联使细胞膜应激得以存活,尽管通过不同的下游效应物。引人注目的是,PKC1 的抑制作用与分子伴侣 Hsp90 或其客户蛋白钙调神经磷酸酶的抑制作用相当。在酿酒酵母中,PKC 信号传导是钙调神经磷酸酶在药物暴露时被激活所必需的。相比之下,在白色念珠菌中,PKc1 和钙调神经磷酸酶通过共同靶点独立调节药物耐药性。我们在白色念珠菌的信号通路中发现了一个额外的调控控制水平,将 PKC 信号、Hsp90 和钙调神经磷酸酶联系起来,因为 Hsp90 的遗传减少导致终端 MAPK,Mkc1 的耗竭。白色念珠菌 PKC1 的缺失使抑菌性麦角固醇生物合成抑制剂具有杀菌作用,并在系统性念珠菌病的小鼠模型中减弱了毒力。这项工作确立了 PKC 信号在耐药性中的新作用,Hsp90 调节耐药性的新通路,以及靶向应激反应信号提供了治疗危及生命的真菌感染的有希望的策略。