Department of Plant Sciences, University of California, Davis, CA 95616, USA.
BMC Genomics. 2010 Dec 14;11:702. doi: 10.1186/1471-2164-11-702.
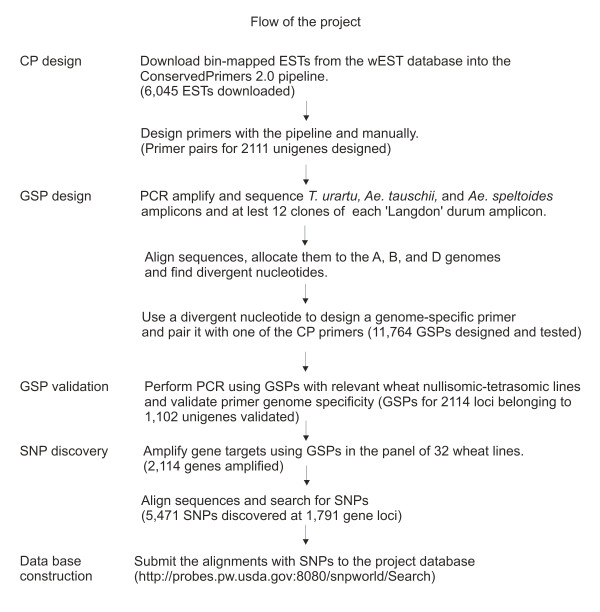
A genome-wide assessment of nucleotide diversity in a polyploid species must minimize the inclusion of homoeologous sequences into diversity estimates and reliably allocate individual haplotypes into their respective genomes. The same requirements complicate the development and deployment of single nucleotide polymorphism (SNP) markers in polyploid species. We report here a strategy that satisfies these requirements and deploy it in the sequencing of genes in cultivated hexaploid wheat (Triticum aestivum, genomes AABBDD) and wild tetraploid wheat (Triticum turgidum ssp. dicoccoides, genomes AABB) from the putative site of wheat domestication in Turkey. Data are used to assess the distribution of diversity among and within wheat genomes and to develop a panel of SNP markers for polyploid wheat.
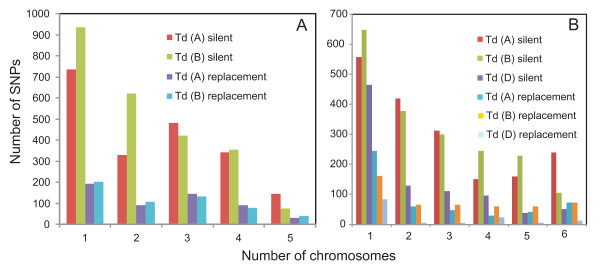
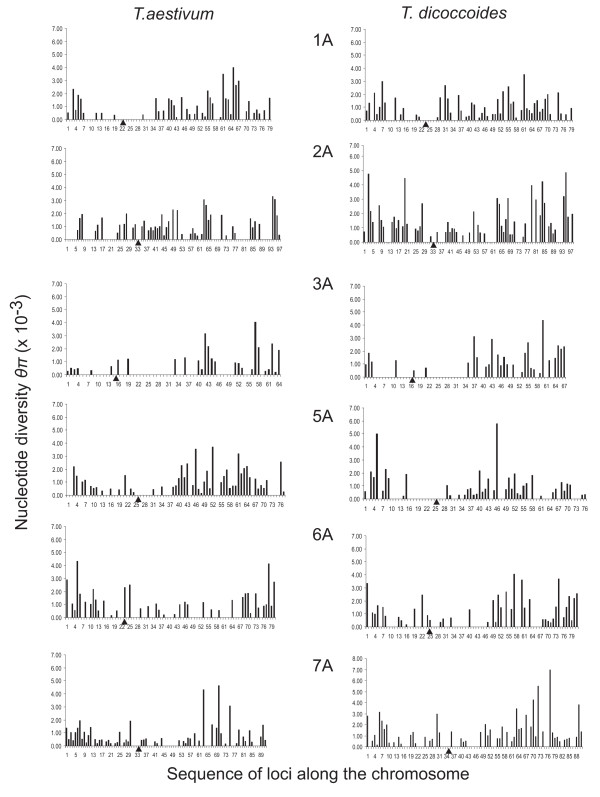
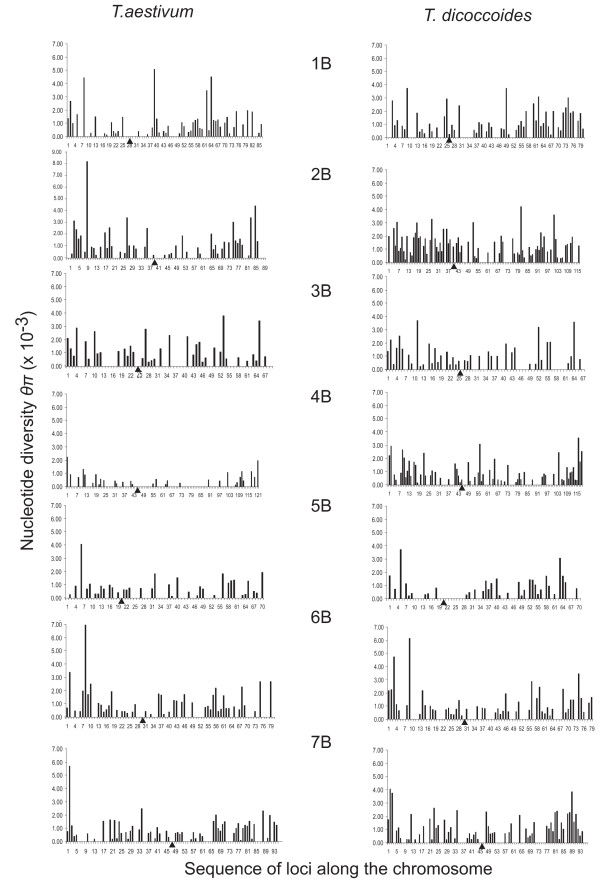
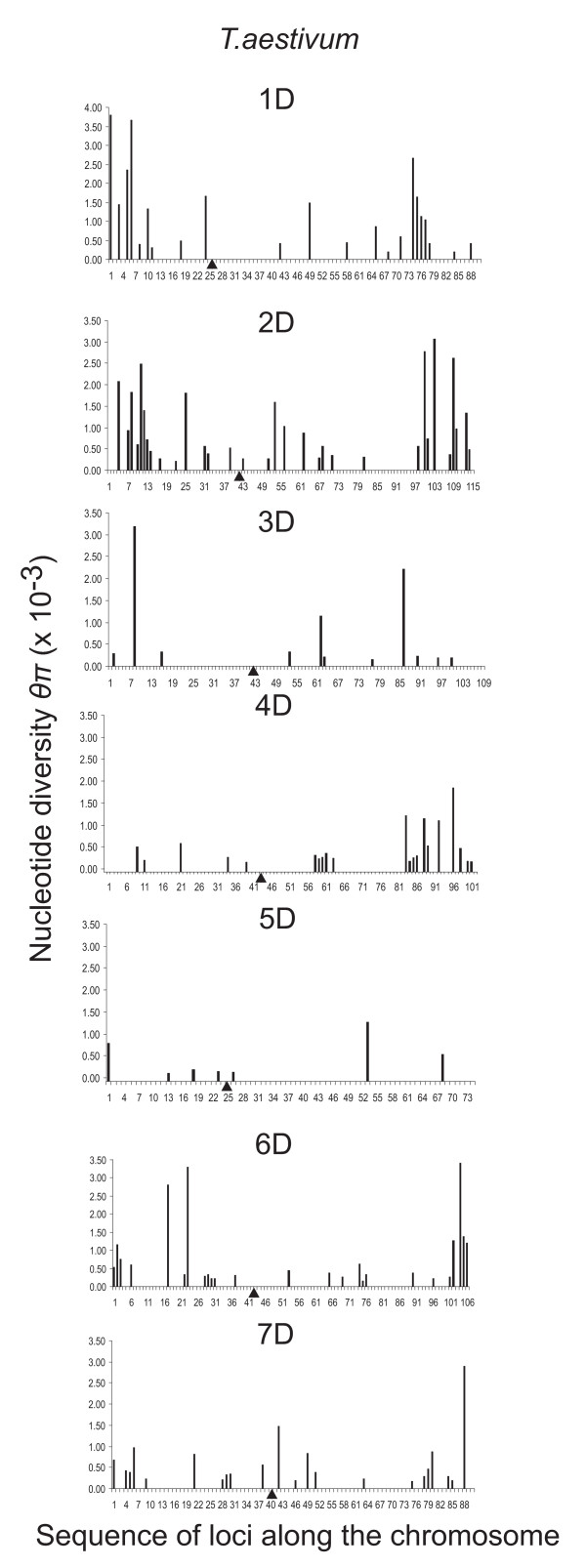
Nucleotide diversity was estimated in 2114 wheat genes and was similar between the A and B genomes and reduced in the D genome. Within a genome, diversity was diminished on some chromosomes. Low diversity was always accompanied by an excess of rare alleles. A total of 5,471 SNPs was discovered in 1791 wheat genes. Totals of 1,271, 1,218, and 2,203 SNPs were discovered in 488, 463, and 641 genes of wheat putative diploid ancestors, T. urartu, Aegilops speltoides, and Ae. tauschii, respectively. A public database containing genome-specific primers, SNPs, and other information was constructed. A total of 987 genes with nucleotide diversity estimated in one or more of the wheat genomes was placed on an Ae. tauschii genetic map, and the map was superimposed on wheat deletion-bin maps. The agreement between the maps was assessed.
In a young polyploid, exemplified by T. aestivum, ancestral species are the primary source of genetic diversity. Low effective recombination due to self-pollination and a genetic mechanism precluding homoeologous chromosome pairing during polyploid meiosis can lead to the loss of diversity from large chromosomal regions. The net effect of these factors in T. aestivum is large variation in diversity among genomes and chromosomes, which impacts the development of SNP markers and their practical utility. Accumulation of new mutations in older polyploid species, such as wild emmer, results in increased diversity and its more uniform distribution across the genome.
在多倍体物种中进行全基因组核苷酸多样性评估时,必须尽量减少同源序列对多样性估计的影响,并可靠地将个体单倍型分配到各自的基因组中。这些相同的要求使在多倍体物种中开发和部署单核苷酸多态性(SNP)标记变得复杂。我们在此报告一种满足这些要求的策略,并将其应用于土耳其被认为是小麦起源地的栽培六倍体小麦(Triticum aestivum,基因组 AABBDD)和野生四倍体小麦(Triticum turgidum ssp. dicoccoides,基因组 AABB)基因的测序中。这些数据用于评估小麦基因组之间和内部的多样性分布,并开发多倍体小麦的 SNP 标记面板。
在 2114 个小麦基因中估计了核苷酸多样性,A 和 B 基因组之间以及 D 基因组中的多样性相似。在一个基因组内,某些染色体上的多样性降低。低多样性总是伴随着稀有等位基因的过剩。在 1791 个小麦基因中发现了 5471 个 SNP。在小麦假定的二倍体祖先 T. urartu、Aegilops speltoides 和 Ae. tauschii 的 488、463 和 641 个基因中分别发现了 1271、1218 和 2203 个 SNP。构建了一个包含基因组特异性引物、SNP 和其他信息的公共数据库。在一个或多个小麦基因组中估计核苷酸多样性的总共 987 个基因被放置在 Ae. tauschii 遗传图谱上,并且该图谱被叠加在小麦缺失-bin 图谱上。评估了图谱之间的一致性。
在以 T. aestivum 为代表的年轻多倍体中,祖先物种是遗传多样性的主要来源。自花授粉导致的有效重组减少以及在多倍体减数分裂中阻止同源染色体配对的遗传机制可能导致来自大染色体区域的多样性丧失。这些因素在 T. aestivum 中的净效应是基因组和染色体之间多样性的巨大差异,这影响了 SNP 标记的开发及其实际应用。在较老的多倍体物种中,如野生二粒小麦,新突变的积累导致多样性增加,并且其在基因组中的分布更加均匀。