Bioinformatics Research Center, Aarhus University, Aarhus, Denmark.
PLoS Genet. 2010 Dec 23;6(12):e1001189. doi: 10.1371/journal.pgen.1001189.
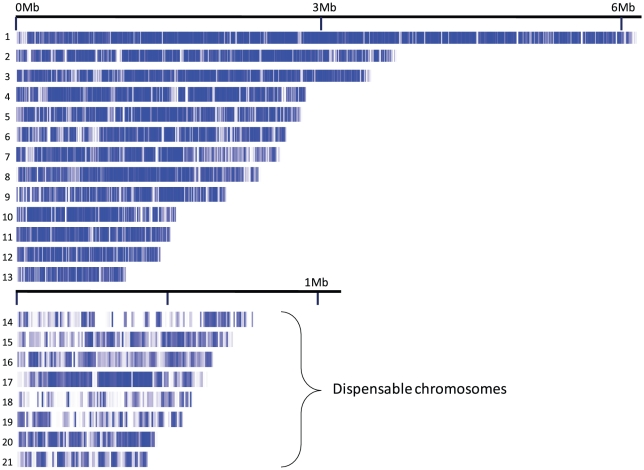
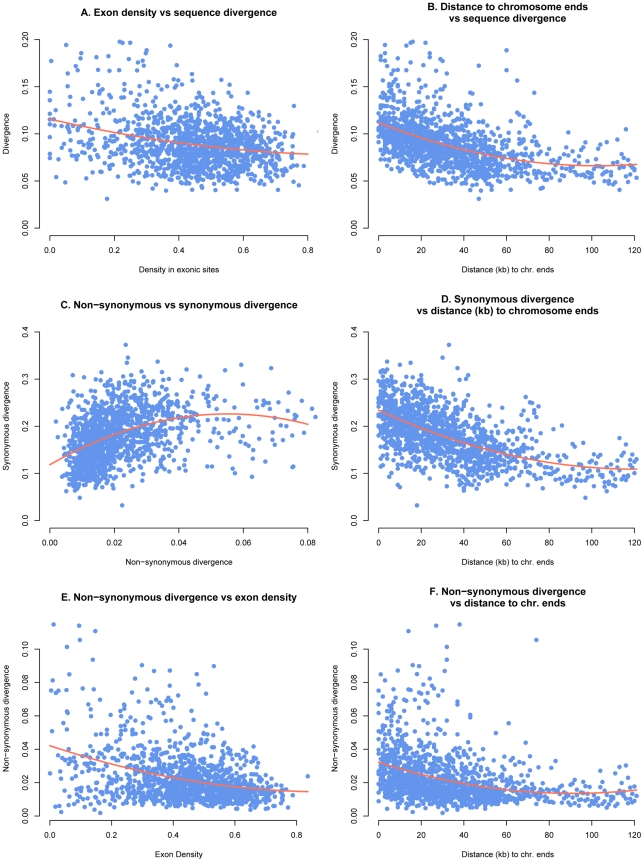
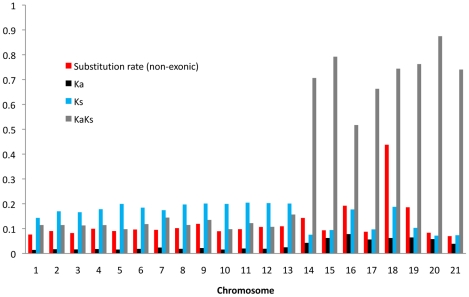
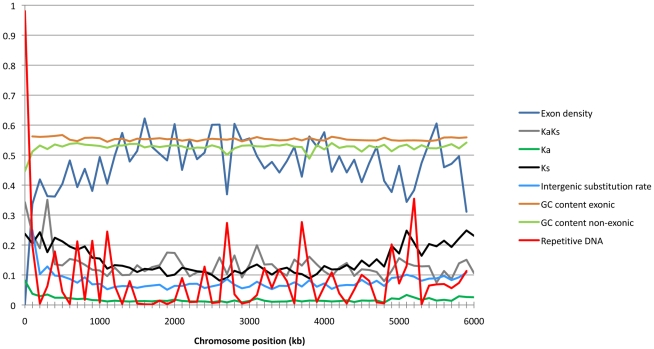
The fungus Mycosphaerella graminicola has been a pathogen of wheat since host domestication 10,000-12,000 years ago in the Fertile Crescent. The wheat-infecting lineage emerged from closely related Mycosphaerella pathogens infecting wild grasses. We use a comparative genomics approach to assess how the process of host specialization affected the genome structure of M. graminicola since divergence from the closest known progenitor species named M. graminicola S1. The genome of S1 was obtained by Illumina sequencing resulting in a 35 Mb draft genome sequence of 32X. Assembled contigs were aligned to the previously sequenced M. graminicola genome. The alignment covered >90% of the non-repetitive portion of the M. graminicola genome with an average divergence of 7%. The sequenced M. graminicola strain is known to harbor thirteen essential chromosomes plus eight dispensable chromosomes. We found evidence that structural rearrangements significantly affected the dispensable chromosomes while the essential chromosomes were syntenic. At the nucleotide level, the essential and dispensable chromosomes have evolved differently. The average synonymous substitution rate in dispensable chromosomes is considerably lower than in essential chromosomes, whereas the average non-synonymous substitution rate is three times higher. Differences in molecular evolution can be related to different transmission and recombination patterns, as well as to differences in effective population sizes of essential and dispensable chromosomes. In order to identify genes potentially involved in host specialization or speciation, we calculated ratios of synonymous and non-synonymous substitution rates in the >9,500 aligned protein coding genes. The genes are generally under strong purifying selection. We identified 43 candidate genes showing evidence of positive selection, one encoding a potential pathogen effector protein. We conclude that divergence of these pathogens was accompanied by structural rearrangements in the small dispensable chromosomes, while footprints of positive selection were present in only a small number of protein coding genes.
真菌禾谷球腔菌自 1 万至 1.2 万年前在新月沃地被人类驯化为小麦的病原体以来,一直是小麦的病原体。感染小麦的谱系是由感染野生草类的密切相关的禾谷球腔菌病原体进化而来。我们使用比较基因组学的方法来评估宿主特化过程自与最接近的已知亲代物种 M.graminicola S1 分化以来,如何影响 M.graminicola 的基因组结构。S1 的基因组是通过 Illumina 测序获得的,产生了一个 35 Mb 的 32X 草图基因组序列。组装的连续序列与之前测序的 M.graminicola 基因组进行比对。比对覆盖了 M.graminicola 基因组非重复部分的 >90%,平均差异为 7%。已知测序的 M.graminicola 菌株含有 13 条必需染色体和 8 条可丢弃染色体。我们发现结构重排显著影响了可丢弃染色体,而必需染色体则是同源的。在核苷酸水平上,必需染色体和可丢弃染色体的进化方式不同。可丢弃染色体的同义替代率明显低于必需染色体,而非同义替代率则高出三倍。分子进化的差异可能与不同的传递和重组模式以及必需和可丢弃染色体的有效种群大小有关。为了鉴定可能参与宿主特化或物种形成的基因,我们计算了在 >9500 个对齐的蛋白质编码基因中同义和非同义替代率的比值。这些基因通常受到强烈的纯化选择。我们鉴定出 43 个候选基因显示出正选择的证据,其中一个基因编码一个潜在的病原体效应蛋白。我们的结论是,这些病原体的分化伴随着小的可丢弃染色体的结构重排,而正选择的痕迹仅存在于少数蛋白质编码基因中。