Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria, 0001, South Africa.
BMC Genomics. 2021 Aug 4;22(1):589. doi: 10.1186/s12864-021-07902-w.
The Botryosphaeriaceae are important plant pathogens, but also have the ability to establish asymptomatic infections that persist for extended periods in a latent state. In this study, we used comparative genome analyses to shed light on the genetic basis of the interactions of these fungi with their plant hosts. For this purpose, we characterised secreted hydrolytic enzymes, secondary metabolite biosynthetic gene clusters and general trends in genomic architecture using all available Botryosphaeriaceae genomes, and selected Dothideomycetes genomes.
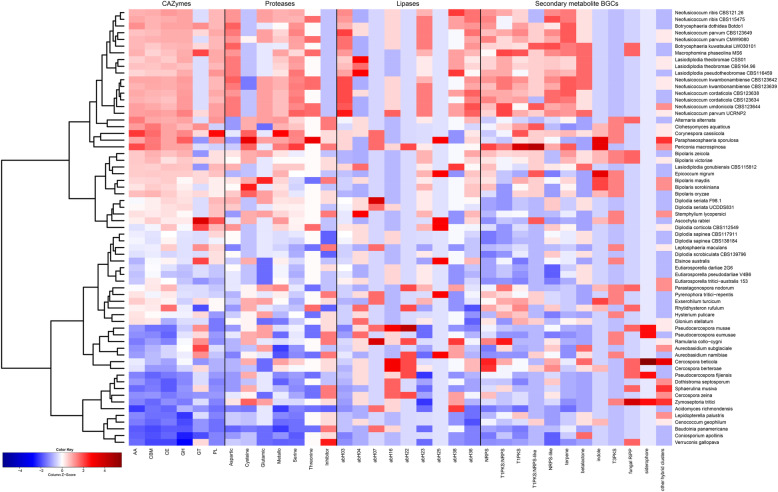
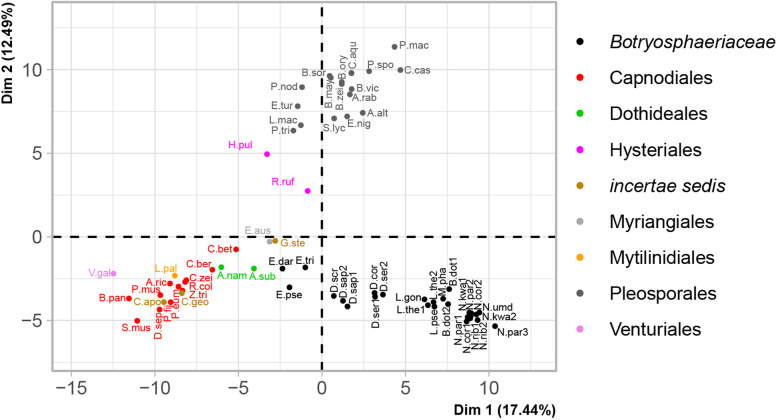
The Botryosphaeriaceae genomes were rich in carbohydrate-active enzymes (CAZymes), proteases, lipases and secondary metabolic biosynthetic gene clusters (BGCs) compared to other Dothideomycete genomes. The genomes of Botryosphaeria, Macrophomina, Lasiodiplodia and Neofusicoccum, in particular, had gene expansions of the major constituents of the secretome, notably CAZymes involved in plant cell wall degradation. The Botryosphaeriaceae genomes were shown to have moderate to high GC contents and most had low levels of repetitive DNA. The genomes were not compartmentalized based on gene and repeat densities, but genes of secreted enzymes were slightly more abundant in gene-sparse regions.
The abundance of secreted hydrolytic enzymes and secondary metabolite BGCs in the genomes of Botryosphaeria, Macrophomina, Lasiodiplodia, and Neofusicoccum were similar to those in necrotrophic plant pathogens and some endophytes of woody plants. The results provide a foundation for comparative genomic analyses and hypotheses to explore the mechanisms underlying Botryosphaeriaceae host-plant interactions.
丛赤壳菌科是重要的植物病原菌,但也具有建立无症状感染的能力,在潜伏状态下持续很长时间。在这项研究中,我们使用比较基因组分析来阐明这些真菌与植物宿主相互作用的遗传基础。为此,我们使用所有可用的丛赤壳菌科基因组和选定的散囊菌目基因组,对分泌水解酶、次生代谢生物合成基因簇和基因组结构的一般趋势进行了特征描述。
与其他散囊菌目基因组相比,丛赤壳菌科基因组富含碳水化合物活性酶(CAZymes)、蛋白酶、脂肪酶和次生代谢生物合成基因簇(BGCs)。特别是,丛赤壳属、大茎点霉属、拟盘多毛孢属和层出镰刀菌属的基因组具有主要分泌成分的基因扩展,特别是参与植物细胞壁降解的 CAZymes。丛赤壳菌科基因组的 GC 含量中等至高,大多数重复 DNA 水平较低。基因组没有根据基因和重复密度进行分隔,但分泌酶的基因在基因稀疏区略为丰富。
丛赤壳属、大茎点霉属、拟盘多毛孢属和层出镰刀菌属基因组中分泌水解酶和次生代谢 BGC 的丰富程度与坏死型植物病原菌和一些木本植物的内生菌相似。这些结果为比较基因组分析和假设提供了基础,以探索丛赤壳菌科与植物宿主相互作用的机制。