Biometris-Applied Statistics, Wageningen University, Wageningen, The Netherlands.
Theor Appl Genet. 2011 May;122(8):1605-16. doi: 10.1007/s00122-011-1558-z. Epub 2011 Mar 4.
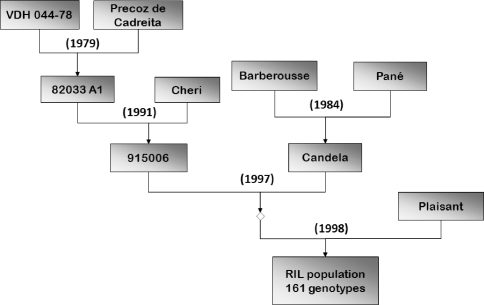
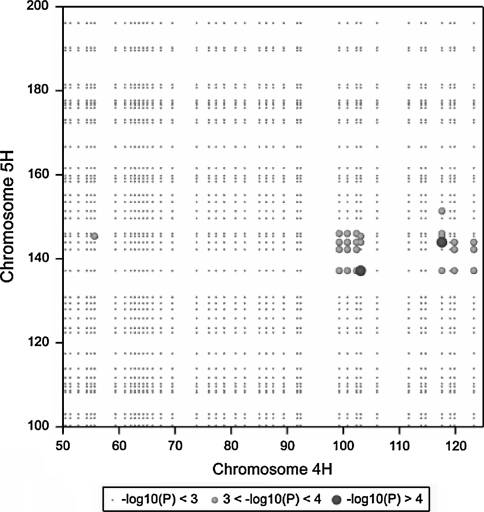
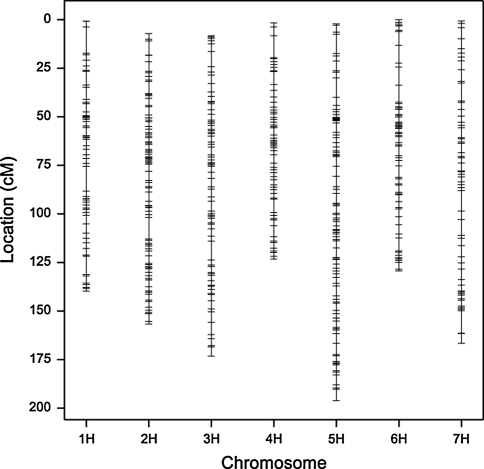
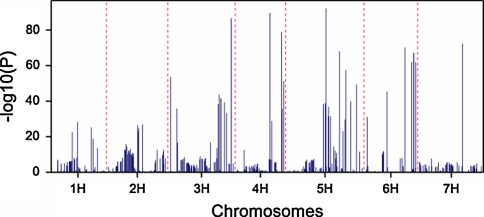
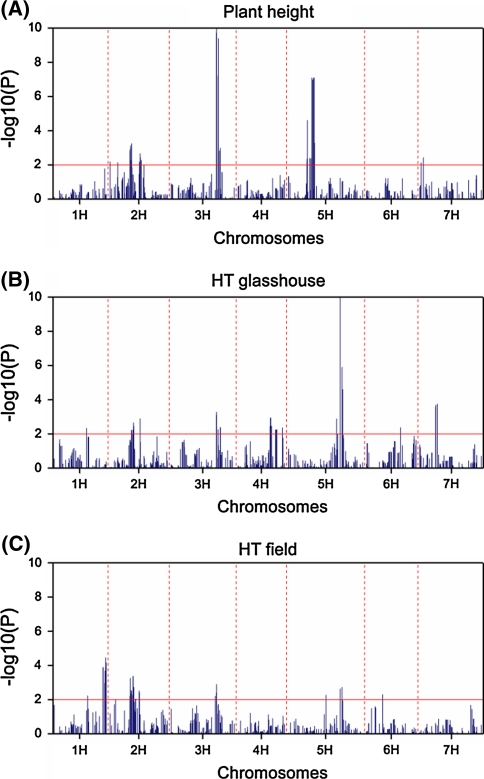
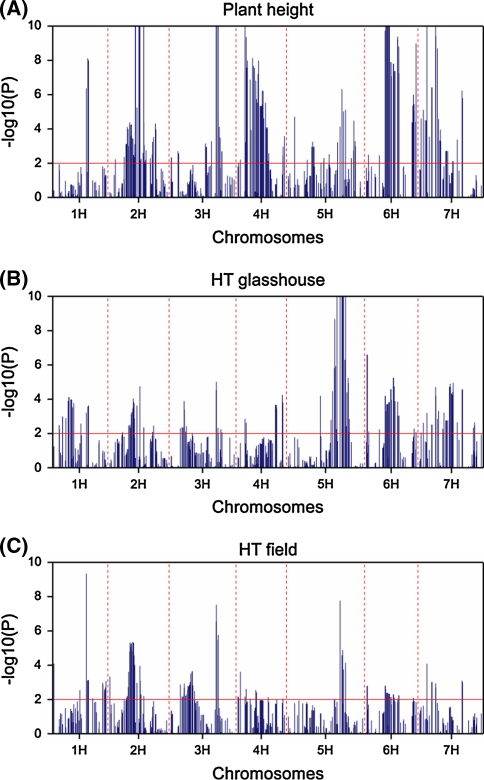
Quantitative trait locus (QTL) detection is commonly performed by analysis of designed segregating populations derived from two inbred parental lines, where absence of selection, mutation and genetic drift is assumed. Even for designed populations, selection cannot always be avoided, with as consequence varying correlation between genotypes instead of uniform correlation. Akin to linkage disequilibrium mapping, ignoring this type of genetic relatedness will increase the rate of false-positives. In this paper, we advocate using mixed models including genetic relatedness, or 'kinship' information for QTL detection in populations where selection forces operated. We demonstrate our case with a three-way barley cross, designed to segregate for dwarfing, vernalization and spike morphology genes, in which selection occurred. The population of 161 inbred lines was screened with 1,536 single nucleotide polymorphisms (SNPs), and used for gene and QTL detection. The coefficient of coancestry matrix was estimated based on the SNPs and imposed to structure the distribution of random genotypic effects. The model incorporating kinship, coancestry, information was consistently superior to the one without kinship (according to the Akaike information criterion). We show, for three traits, that ignoring the coancestry information results in an unrealistically high number of marker-trait associations, without providing clear conclusions about QTL locations. We used a number of widely recognized dwarfing and vernalization genes known to segregate in the studied population as landmarks or references to assess the agreement of the mapping results with a priori candidate gene expectations. Additional QTLs to the major genes were detected for all traits as well.
数量性状位点(QTL)检测通常通过分析来自两个近交亲本系的设计分离群体进行,假设不存在选择、突变和遗传漂变。即使是对于设计的群体,选择也不能总是避免,其结果是基因型之间的相关性不同,而不是均匀的相关性。与连锁不平衡作图类似,忽略这种类型的遗传相关性会增加假阳性的比率。在本文中,我们主张在存在选择力的群体中使用包含遗传相关性或“亲缘关系”信息的混合模型进行 QTL 检测。我们用一个三向大麦杂交设计来说明我们的案例,该设计旨在分离矮化、春化和穗形态基因,其中发生了选择。161 个近交系的群体用 1536 个单核苷酸多态性(SNP)进行了筛选,并用于基因和 QTL 检测。根据 SNP 估计了亲缘关系系数矩阵,并将其用于构建随机基因型效应的分布。包含亲缘关系、亲缘关系信息的模型始终优于没有亲缘关系的模型(根据 Akaike 信息准则)。我们展示了三个性状,忽略亲缘关系信息会导致标记-性状关联的数量不切实际地增加,而没有提供关于 QTL 位置的明确结论。我们使用了一些广泛认可的在研究群体中分离的矮化和春化基因作为地标或参考,以评估映射结果与先验候选基因预期的一致性。所有性状都检测到了主要基因的其他 QTL。