Program in Molecular and Computational Biology, University of Southern California, Los Angeles, CA 90089, USA.
Bioinformatics. 2011 Jul 1;27(13):i69-76. doi: 10.1093/bioinformatics/btr207.
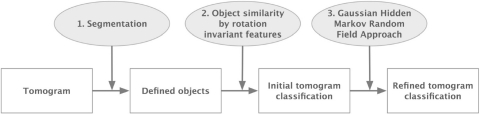
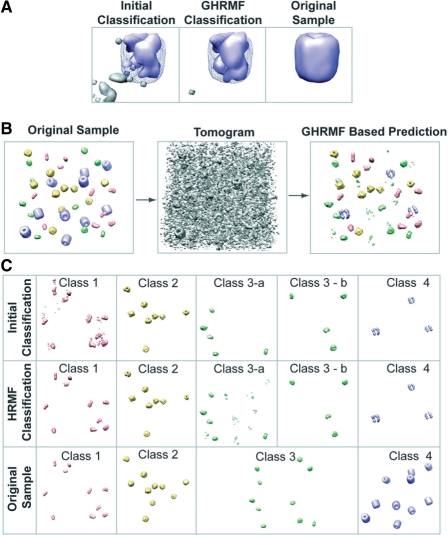
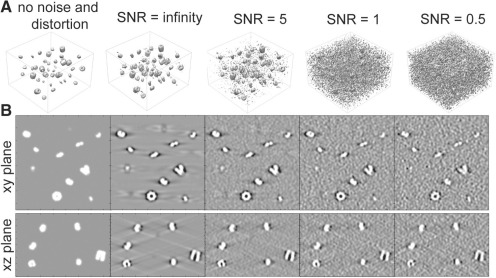
Cryo electron tomography (CryoET) produces 3D density maps of biological specimen in its near native states. Applied to small cells, cryoET produces 3D snapshots of the cellular distributions of large complexes. However, retrieving this information is non-trivial due to the low resolution and low signal-to-noise ratio in tomograms. Current pattern recognition methods identify complexes by matching known structures to the cryo electron tomogram. However, so far only a small fraction of all protein complexes have been structurally resolved. It is, therefore, of great importance to develop template-free methods for the discovery of previously unknown protein complexes in cryo electron tomograms.
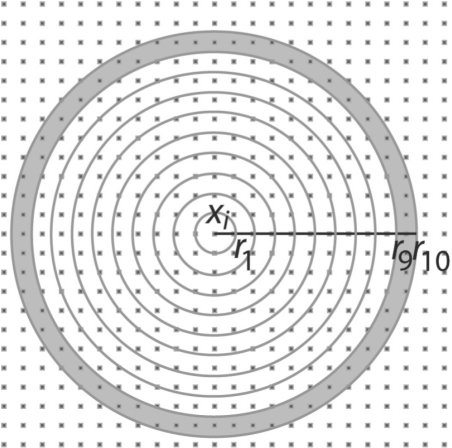
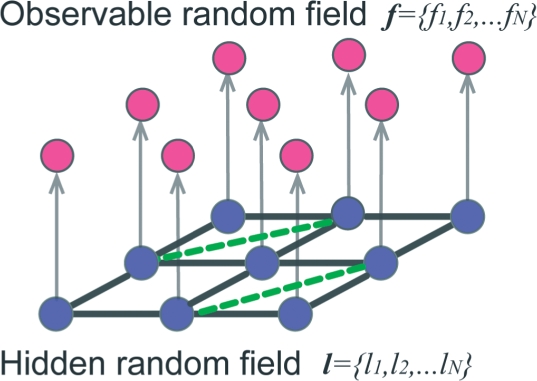
Here, we have developed an inference method for the template-free discovery of frequently occurring protein complexes in cryo electron tomograms. We provide a first proof-of-principle of the approach and assess its applicability using realistically simulated tomograms, allowing for the inclusion of noise and distortions due to missing wedge and electron optical factors. Our method is a step toward the template-free discovery of the shapes, abundance and spatial distributions of previously unknown macromolecular complexes in whole cell tomograms.
低温电子断层扫描(CryoET)以其近天然状态产生生物样本的 3D 密度图。应用于小细胞,低温电子断层扫描产生了大复合物在细胞内分布的 3D 快照。然而,由于断层图像的低分辨率和低信噪比,检索这些信息并非易事。目前的模式识别方法通过将已知结构与低温电子断层扫描匹配来识别复合物。然而,到目前为止,只有一小部分蛋白质复合物的结构已经得到解决。因此,开发无模板方法来发现低温电子断层扫描中以前未知的蛋白质复合物具有重要意义。
在这里,我们开发了一种无模板方法,用于在低温电子断层扫描中发现经常出现的蛋白质复合物。我们提供了该方法的第一个原理证明,并使用真实模拟的断层扫描评估了其适用性,允许包括由于缺失楔形和电子光学因素引起的噪声和失真。我们的方法是朝着在整个细胞断层扫描中发现以前未知的大分子复合物的形状、丰度和空间分布的无模板发现迈出的一步。