Institut Pasteur, CIBU, Department Infection and Epidemiology, Paris, France.
Infect Genet Evol. 2011 Oct;11(7):1690-702. doi: 10.1016/j.meegid.2011.06.021. Epub 2011 Jul 8.
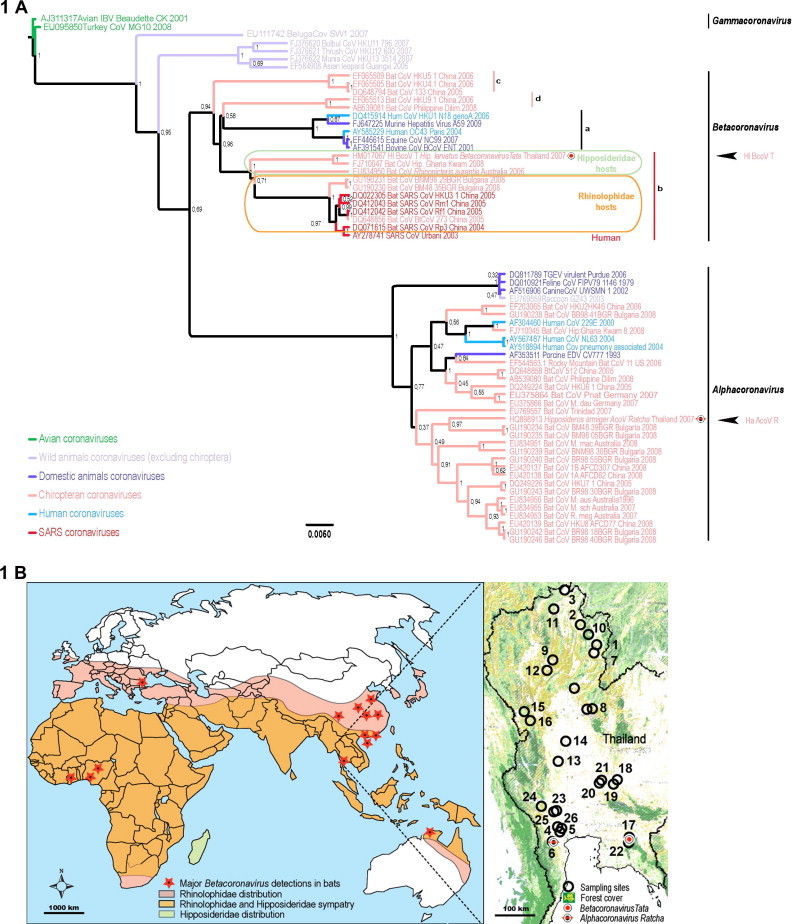
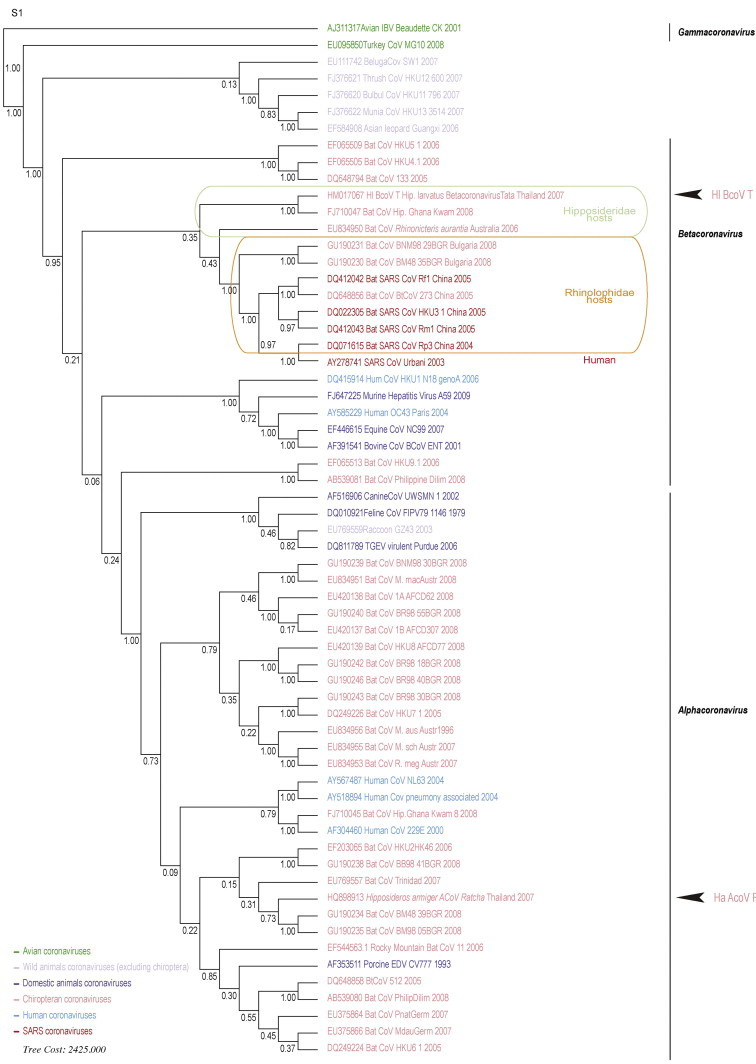
One of the great challenges in the ecology of infectious diseases is to understand what drives the emergence of new pathogens including the relationship between viruses and their hosts. In the case of the emergence of SevereAcute Respiratory Syndrome Coronavirus (SARS-CoV), several studies have shown coronavirus diversity in bats as well as the existence of SARS-CoV infection in apparently healthy bats, suggesting that bats may be a crucial host in the genesis of this disease. To elucidate the biogeographic origin of SARS-CoV and investigate the role that bats played in its emergence, we amplified coronavirus sequences from bat species captured throughout Thailand and assessed the phylogenetic relationships to each other and to other published coronavirus sequences. To this end, RdRp sequence of Coronavirinae was targeted by RT-PCR in non-invasive samples from bats collected in Thailand. Two new coronaviruses were detected in two bat species: one Betacoronavirus in Hipposideros larvatus and one Alphacoronavirus in Hipposiderosarmiger. Interestingly, these viruses from South-East Asia are related to those previously detected in Africa (Betacoronavirus-b) or in Europe (Alphacoronavirus & Betacoronavirus-b). These findings illuminate the origin and the evolutionary history of the SARS-CoV group found in bats by pushing forward the hypothesis of a Betacoronavirus spill-over from Hipposideridae to Rhinolophidae and then from Rhinolophidae to civets and Human. All reported Betacoronaviruses-b (SARS-CoV group) of Hipposideridae and Rhinolophidae respectively cluster in two groups despite their broad geographic distribution and the sympatry of their hosts, which is in favor of an ancient and genetically independent evolution of Betacoronavirus-b clusters in these families. Moreover, despite its probable pathogenicity, we found that a Betacoronavirus-b can persistently infect a medium-sized hipposiderid bat colony. These findings illustrate the importance of the host phylogeny and the host/pathogen ecological interactions in the description and the understanding of pathogen emergence. The host's phylogeny, biogeography and behaviour, combined with already described roles of pathogen plasticity and anthropic changes are likely to be co-factors of disease emergence. Elucidating the common ancestor of Hipposideridae and Rhinolophidae is key to understanding the evolutionary history of actual betacoronaviruses and therefore to get an insight of the deep origin of SARS-CoV.
传染病生态学的一大挑战是要了解是什么驱动了新病原体的出现,包括病毒与其宿主之间的关系。就严重急性呼吸系统综合症冠状病毒(SARS-CoV)的出现而言,几项研究表明蝙蝠体内冠状病毒多样性以及在显然健康的蝙蝠中存在 SARS-CoV 感染,这表明蝙蝠可能是这种疾病起源的关键宿主。为了阐明 SARS-CoV 的生物地理起源并研究蝙蝠在其出现中所扮演的角色,我们从泰国各地捕获的蝙蝠物种中扩增了冠状病毒序列,并评估了彼此之间以及与其他已发表的冠状病毒序列的系统发育关系。为此,我们通过 RT-PCR 从泰国采集的蝙蝠非侵入性样本中靶向冠状病毒科 RdRp 序列。在两种蝙蝠物种中检测到两种新的冠状病毒:一种在 Hipposideros larvatus 中的β冠状病毒和一种在 Hipposiderosarmiger 中的α冠状病毒。有趣的是,这些来自东南亚的病毒与先前在非洲(β冠状病毒-b)或欧洲(α冠状病毒和β冠状病毒-b)检测到的病毒有关。这些发现通过推进从 Hipposideridae 到 Rhinolophidae 的β冠状病毒溢出的假说,并进一步从 Rhinolophidae 到果子狸和人类,阐明了蝙蝠中 SARS-CoV 组的起源和进化历史。分别来自 Hipposideridae 和 Rhinolophidae 的所有报告的β冠状病毒-b(SARS-CoV 组)尽管具有广泛的地理分布和宿主的同域性,但仍分别聚类成两个组,这有利于这两个科的β冠状病毒-b 聚类的古老和遗传独立性进化。此外,尽管可能具有致病性,但我们发现一种β冠状病毒-b 可以持续感染中型 Hipposideridae 蝙蝠群。这些发现说明了宿主系统发育和宿主/病原体生态相互作用在描述和理解病原体出现方面的重要性。宿主的系统发育、生物地理学和行为,结合已经描述的病原体可塑性和人为变化的作用,可能是疾病出现的共同因素。阐明 Hipposideridae 和 Rhinolophidae 的共同祖先对于了解实际的β冠状病毒的进化历史至关重要,因此可以深入了解 SARS-CoV 的起源。