Department of Biomedical Informatics, University of Pittsburgh, Pittsburgh, Pennsylvania, United States of America.
PLoS One. 2011;6(8):e22075. doi: 10.1371/journal.pone.0022075. Epub 2011 Aug 10.
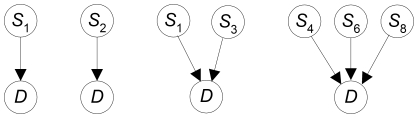
A genome-wide association study (GWAS) typically involves examining representative SNPs in individuals from some population. A GWAS data set can concern a million SNPs and may soon concern billions. Researchers investigate the association of each SNP individually with a disease, and it is becoming increasingly commonplace to also analyze multi-SNP associations. Techniques for handling so many hypotheses include the Bonferroni correction and recently developed bayesian methods. These methods can encounter problems. Most importantly, they are not applicable to a complex multi-locus hypothesis which has several competing hypotheses rather than only a null hypothesis. A method that computes the posterior probability of complex hypotheses is a pressing need.
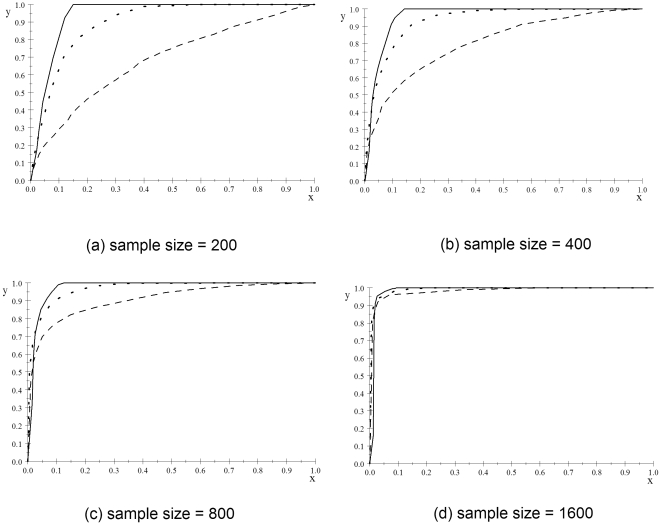
METHODOLOGY/FINDINGS: We introduce the bayesian network posterior probability (BNPP) method which addresses the difficulties. The method represents the relationship between a disease and SNPs using a directed acyclic graph (DAG) model, and computes the likelihood of such models using a bayesian network scoring criterion. The posterior probability of a hypothesis is computed based on the likelihoods of all competing hypotheses. The BNPP can not only be used to evaluate a hypothesis that has previously been discovered or suspected, but also to discover new disease loci associations. The results of experiments using simulated and real data sets are presented. Our results concerning simulated data sets indicate that the BNPP exhibits both better evaluation and discovery performance than does a p-value based method. For the real data sets, previous findings in the literature are confirmed and additional findings are found.
CONCLUSIONS/SIGNIFICANCE: We conclude that the BNPP resolves a pressing problem by providing a way to compute the posterior probability of complex multi-locus hypotheses. A researcher can use the BNPP to determine the expected utility of investigating a hypothesis further. Furthermore, we conclude that the BNPP is a promising method for discovering disease loci associations.
全基因组关联研究(GWAS)通常涉及检查来自某些人群的个体中的代表性 SNP。GWAS 数据集可能涉及一百万 SNP,并且很快可能涉及数十亿 SNP。研究人员单独研究每个 SNP 与疾病的关联,并且分析多 SNP 关联也越来越普遍。处理如此多假设的技术包括 Bonferroni 校正和最近开发的贝叶斯方法。这些方法可能会遇到问题。最重要的是,它们不适用于具有多个竞争假设而不是仅一个零假设的复杂多基因座假设。计算复杂假设后验概率的方法是迫切需要的。
方法/发现:我们引入了贝叶斯网络后验概率(BNPP)方法来解决这些困难。该方法使用有向无环图(DAG)模型表示疾病与 SNPs 之间的关系,并使用贝叶斯网络评分标准计算此类模型的可能性。根据所有竞争假设的可能性来计算假设的后验概率。BNPP 不仅可用于评估先前已发现或怀疑的假设,还可用于发现新的疾病相关位点。使用模拟和真实数据集的实验结果。关于模拟数据集的结果表明,BNPP 不仅在评估性能方面表现更好,而且在发现性能方面也表现更好。对于真实数据集,确认了文献中的先前发现,并发现了其他发现。
结论/意义:我们得出结论,BNPP 通过提供一种计算复杂多基因座假设后验概率的方法解决了一个紧迫的问题。研究人员可以使用 BNPP 进一步确定调查假设的预期效用。此外,我们得出结论,BNPP 是发现疾病相关位点关联的一种很有前途的方法。