Southern California Research Centers for Alcoholic Liver and Pancreatic Diseases and Cirrhosis, VA Greater Los Angeles Healthcare System, and University of California at Los Angeles, California 90073, USA.
J Gastroenterol Hepatol. 2012 Mar;27 Suppl 2(Suppl 2):27-32. doi: 10.1111/j.1440-1746.2011.07004.x.
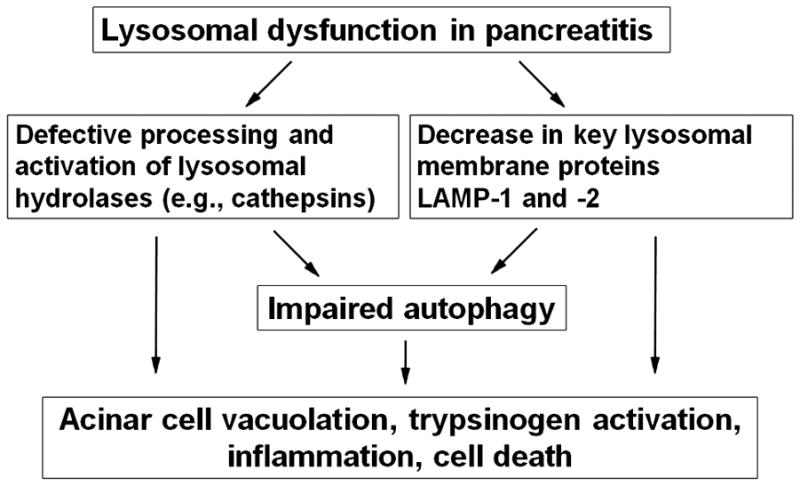
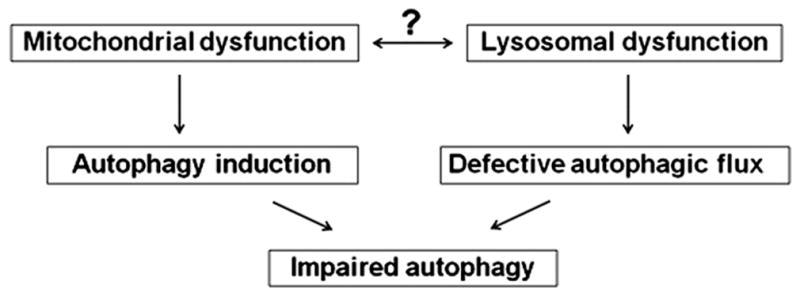
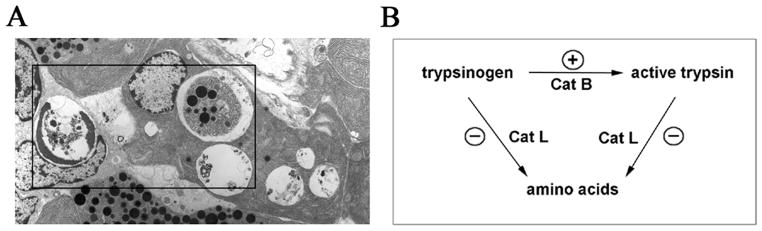
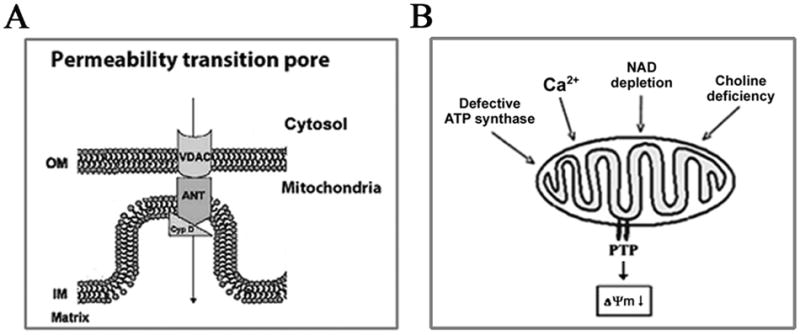
Recent findings from our group, obtained on experimental in vivo and ex vivo models of pancreatitis, reveal that this disease causes a profound dysfunction of key cellular organelles, lysosomes and mitochondria. We found that autophagy, the main cellular degradative, lysosome-driven process, is activated but also impaired in acute pancreatitis because of its' inefficient progression/resolution (flux) resulting from defective function of lysosomes. One mechanism underlying the lysosomal dysfunction in pancreatitis is abnormal processing (maturation) and activation of cathepsins, major lysosomal hydrolases; another is a decrease in pancreatic levels of key lysosomal membrane proteins LAMP-1 and LAMP-2. Our data indicate that lysosomal dysfunction plays an important initiating role in pancreatitis pathobiology. The impaired autophagy mediates vacuole accumulation in acinar cells; furthermore, the abnormal maturation and activation of cathepsins leads to increase in intra-acinar trypsin, the hallmark of pancreatitis; and LAMP-2 deficiency causes inflammation and acinar cell necrosis. Thus, the autophagic and lysosomal dysfunctions mediate key pathologic responses of pancreatitis. On the other hand, we showed that pancreatitis causes acinar cell mitochondria depolarization, mediated by the permeability transition pore (PTP). Genetic (via deletion of cyclophilin D) inactivation of PTP prevents mitochondrial depolarization and greatly ameliorates the pathologic responses of pancreatitis. Further, our data suggest that mitochondrial damage, by stimulating autophagy, increases the demand for efficient lysosomal degradation and therefore aggravates the pathologic consequences of lysosomal dysfunction. Thus, the combined autophagic, lysosomal and mitochondrial dysfunctions are key to the pathogenesis of pancreatitis.
我们小组最近在胰腺炎的实验体内和体外模型中发现,这种疾病会导致关键细胞细胞器(溶酶体和线粒体)严重功能障碍。我们发现,自噬是主要的细胞降解、溶酶体驱动的过程,在急性胰腺炎中被激活,但也受到损害,因为其进展/解决(通量)效率低下,原因是溶酶体功能缺陷。胰腺炎中溶酶体功能障碍的一个机制是组织蛋白酶的异常加工(成熟)和激活,组织蛋白酶是主要的溶酶体水解酶;另一个机制是关键溶酶体膜蛋白 LAMP-1 和 LAMP-2 在胰腺中的水平下降。我们的数据表明,溶酶体功能障碍在胰腺炎的病理生物学中起着重要的起始作用。受损的自噬介导了上皮细胞中的空泡积累;此外,组织蛋白酶的异常成熟和激活导致胰蛋白酶在腺泡内增加,这是胰腺炎的标志;LAMP-2 缺乏导致炎症和上皮细胞坏死。因此,自噬和溶酶体功能障碍介导了胰腺炎的关键病理反应。另一方面,我们表明胰腺炎导致上皮细胞线粒体去极化,这是由通透性转换孔(PTP)介导的。PTP 的遗传失活(通过删除亲环素 D)可防止线粒体去极化,并极大地改善胰腺炎的病理反应。此外,我们的数据表明,线粒体损伤通过刺激自噬,增加了对有效溶酶体降解的需求,从而加剧了溶酶体功能障碍的病理后果。因此,自噬、溶酶体和线粒体功能障碍的联合是胰腺炎发病机制的关键。