Department of Genome Sciences, University of Washington, Seattle, Washington, USA.
mBio. 2012 Mar 20;3(2). doi: 10.1128/mBio.00001-12. Print 2012.
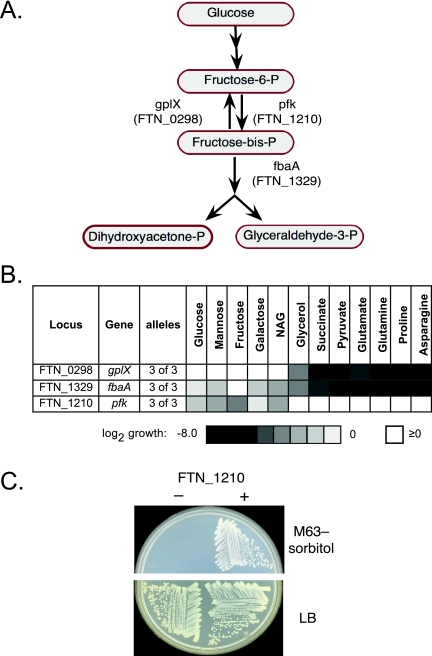
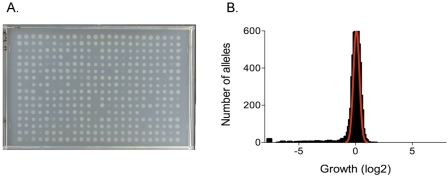
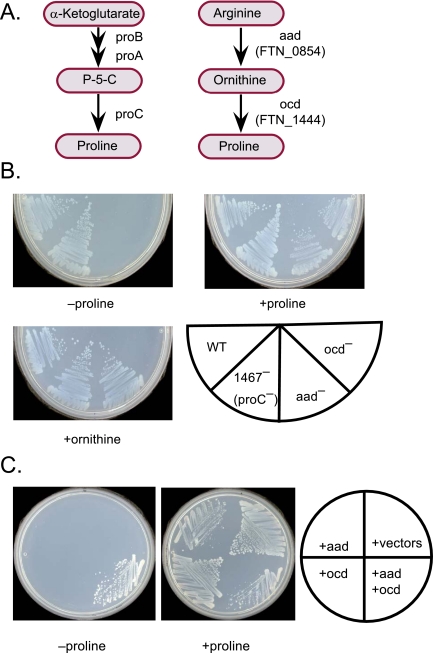
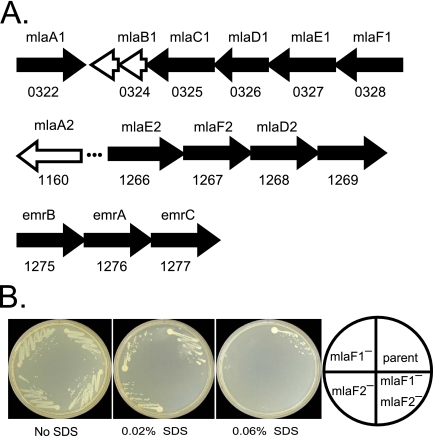
To help define the biological functions of nonessential genes of Francisella novicida, we measured the growth of arrayed members of a comprehensive transposon mutant library under a variety of nutrition and stress conditions. Mutant phenotypes were identified for 37% of the genes, corresponding to ten carbon source utilization pathways, nine amino acid- and nucleotide-biosynthetic pathways, ten intrinsic antibiotic resistance traits, and six other stress resistance traits. The greatest surprise of the analysis was the large number of genotype-phenotype relationships that were not predictable from studies of Escherichia coli and other model species. The study identified candidate genes for a missing glycolysis function (phosphofructokinase), an unusual proline-biosynthetic pathway, parallel outer membrane lipid asymmetry maintenance systems, and novel antibiotic resistance functions. The analysis provides an evaluation of annotation predictions, identifies cases in which fundamental processes differ from those in model species, and helps create an empirical foundation for understanding virulence and other complex processes.
The value of genome sequences as foundations for analyzing complex traits in nonmodel organisms is limited by the need to rely almost exclusively on sequence similarities to predict gene functions in annotations. Many genes cannot be assigned functions, and some predictions are incorrect or incomplete. Due to these limitations, genome-scale experimental approaches that test and extend bioinformatics-based predictions are sorely needed. In this study, we describe such an approach based on phenotypic analysis of a comprehensive, sequence-defined transposon mutant library.
为了帮助确定弗氏柠檬酸杆菌非必需基因的生物学功能,我们在多种营养和应激条件下测量了综合转座子突变体文库中排列成员的生长情况。鉴定出 37%的基因的突变表型,对应十种碳源利用途径、九种氨基酸和核苷酸生物合成途径、十种固有抗生素抗性特性和六种其他应激抗性特性。分析中最令人惊讶的是,大量的基因型-表型关系无法从大肠杆菌和其他模式物种的研究中预测出来。该研究确定了缺失的糖酵解功能(磷酸果糖激酶)、不寻常的脯氨酸生物合成途径、平行的外膜脂质不对称维持系统和新型抗生素抗性功能的候选基因。该分析评估了注释预测的准确性,确定了基本过程与模型物种不同的情况,并有助于为理解毒力和其他复杂过程创建一个经验基础。
基因组序列作为分析非模式生物复杂特征的基础的价值受到需要几乎完全依赖于序列相似性来预测注释中基因功能的限制。许多基因无法被赋予功能,有些预测是错误或不完整的。由于这些限制,急需基于实验方法的基因组规模的方法来测试和扩展基于生物信息学的预测。在这项研究中,我们描述了一种基于综合、序列定义的转座子突变体文库的表型分析的方法。