Blazier Anna S, Papin Jason A
Department of Biomedical Engineering, University of Virginia, Charlottesville VA, USA.
Front Physiol. 2012 Aug 6;3:299. doi: 10.3389/fphys.2012.00299. eCollection 2012.
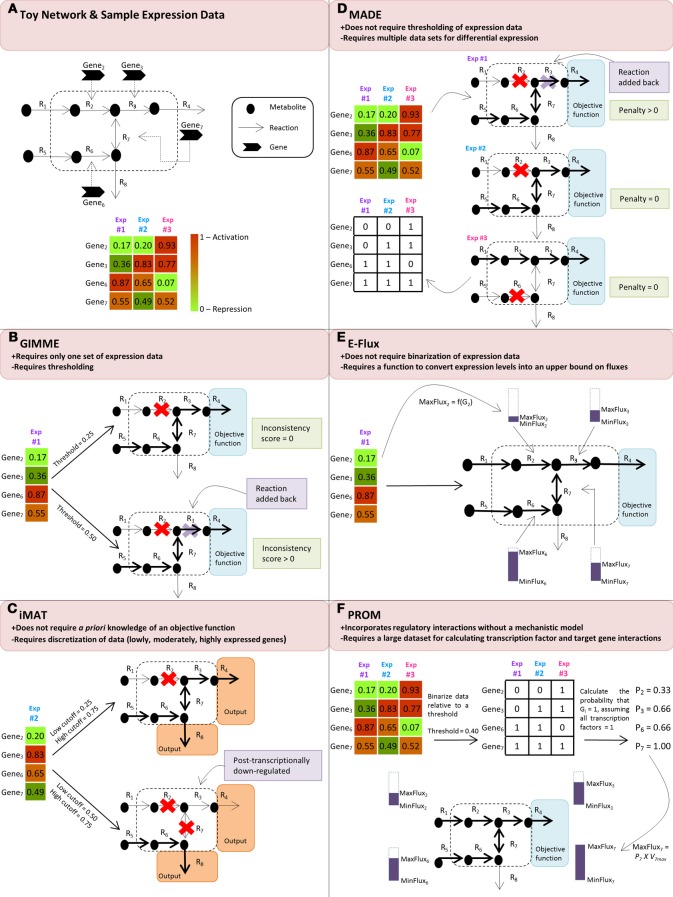
With the advent of high-throughput technologies, the field of systems biology has amassed an abundance of "omics" data, quantifying thousands of cellular components across a variety of scales, ranging from mRNA transcript levels to metabolite quantities. Methods are needed to not only integrate this omics data but to also use this data to heighten the predictive capabilities of computational models. Several recent studies have successfully demonstrated how flux balance analysis (FBA), a constraint-based modeling approach, can be used to integrate transcriptomic data into genome-scale metabolic network reconstructions to generate predictive computational models. In this review, we summarize such FBA-based methods for integrating expression data into genome-scale metabolic network reconstructions, highlighting their advantages as well as their limitations.
随着高通量技术的出现,系统生物学领域积累了大量的“组学”数据,这些数据在从mRNA转录水平到代谢物数量等各种尺度上对数千种细胞成分进行了量化。不仅需要整合这些组学数据的方法,还需要利用这些数据来提高计算模型的预测能力。最近的几项研究成功地证明了通量平衡分析(FBA),一种基于约束的建模方法,如何能够用于将转录组数据整合到基因组规模的代谢网络重建中,以生成预测性计算模型。在这篇综述中,我们总结了基于FBA的将表达数据整合到基因组规模代谢网络重建中的方法,突出了它们的优点和局限性。