Département d'Informatique, DIRO, Université de Montréal, Canada.
BMC Bioinformatics. 2012;13 Suppl 19(Suppl 19):S4. doi: 10.1186/1471-2105-13-S19-S4. Epub 2012 Dec 19.
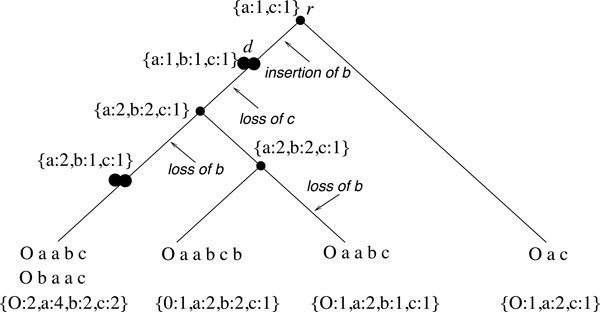
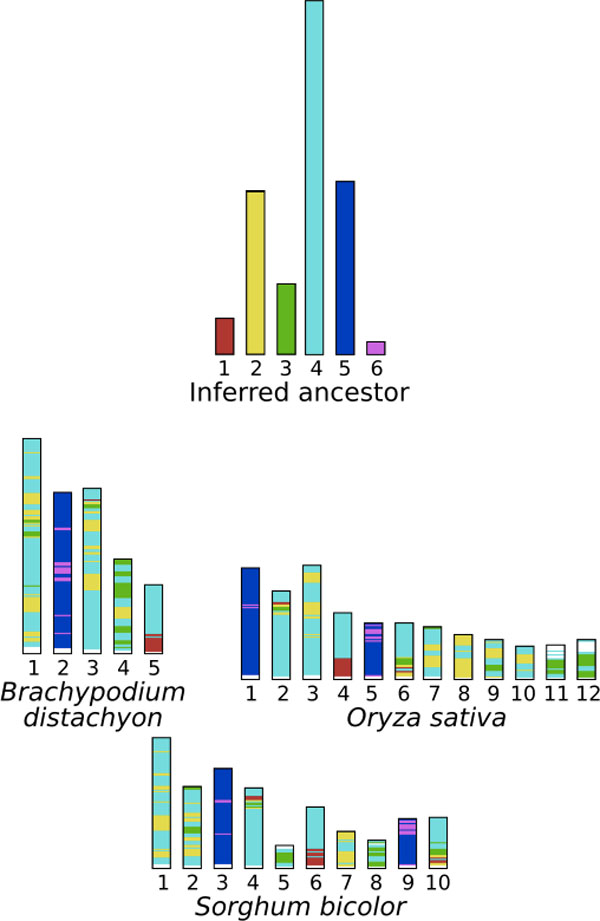
The "small phylogeny" problem consists in inferring ancestral genomes associated with each internal node of a phylogenetic tree of a set of extant species. Existing methods can be grouped into two main categories: the distance-based methods aiming at minimizing a total branch length, and the synteny-based (or mapping) methods that first predict a collection of relations between ancestral markers in term of "synteny", and then assemble this collection into a set of Contiguous Ancestral Regions (CARs). The predicted CARs are likely to be more reliable as they are more directly deduced from observed conservations in extant species. However the challenge is to end up with a completely assembled genome.
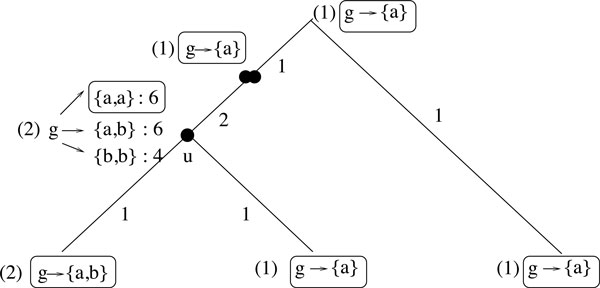
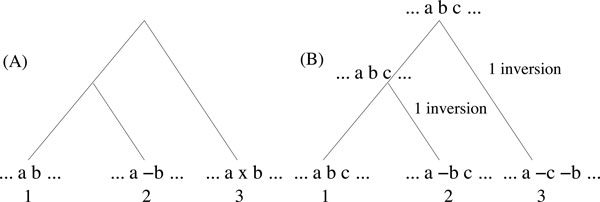
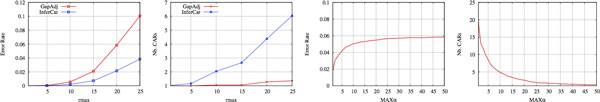
We develop a new synteny-based method that is flexible enough to handle a model of evolution involving whole genome duplication events, in addition to rearrangements, gene insertions, and losses. Ancestral relationships between markers are defined in term of Gapped Adjacencies, i.e. pairs of markers separated by up to a given number of markers. It improves on a previous restricted to direct adjacencies, which revealed a high accuracy for adjacency prediction, but with the drawback of being overly conservative, i.e. of generating a large number of CARs. Applying our algorithm on various simulated data sets reveals good performance as we usually end up with a completely assembled genome, while keeping a low error rate.
All source code is available at http://www.iro.umontreal.ca/~mabrouk.
“小系统发生”问题在于推断与系统发生树中每个内部节点相关的祖先基因组。现有的方法可以分为两大类:基于距离的方法旨在最小化总分支长度,以及基于同线性(或映射)的方法,这些方法首先预测在“同线性”方面祖先标记之间的关系集合,然后将该集合组装成一组连续的祖先区域(CAR)。由于它们更直接地从现存物种中的保守性推断出来,因此预测的 CAR 可能更可靠。然而,挑战在于最终得到一个完全组装的基因组。
我们开发了一种新的基于同线性的方法,它足够灵活,可以处理涉及全基因组复制事件的进化模型,以及重排、基因插入和缺失。标记之间的祖先关系是根据有缺口的邻接来定义的,即最多由给定数量的标记分隔的一对标记。它改进了以前仅限于直接邻接的方法,该方法在邻接预测方面具有很高的准确性,但缺点是过于保守,即产生大量的 CAR。我们的算法在各种模拟数据集上的应用显示出了良好的性能,因为我们通常最终得到一个完全组装的基因组,同时保持低错误率。
所有的源代码都可以在 http://www.iro.umontreal.ca/~mabrouk. 上获得。