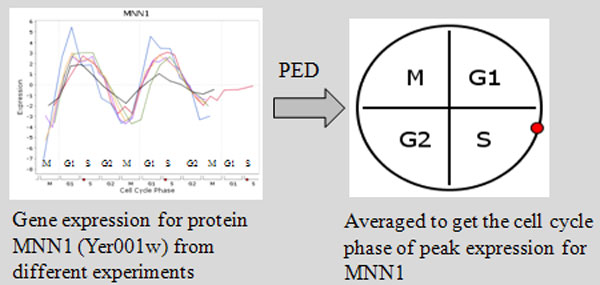
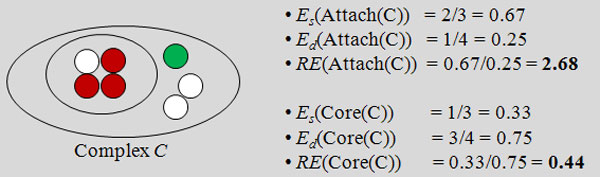
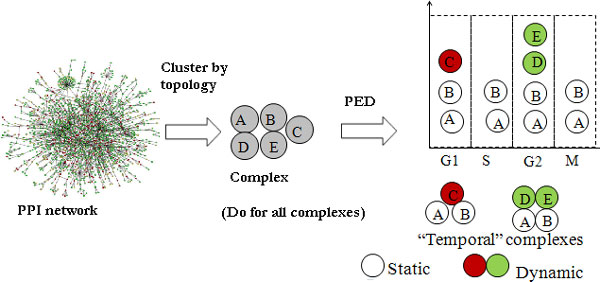
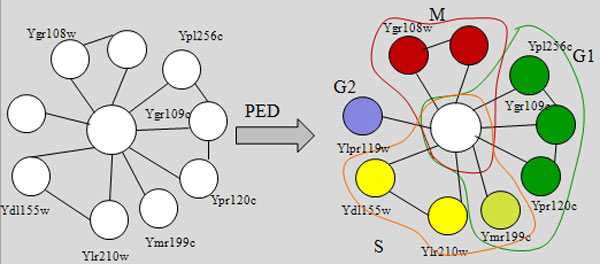
Department of Computer Science, National University of Singapore, Singapore.
BMC Bioinformatics. 2012;13 Suppl 17(Suppl 17):S16. doi: 10.1186/1471-2105-13-S17-S16. Epub 2012 Dec 13.
Complexes of physically interacting proteins are one of the fundamental functional units responsible for driving key biological mechanisms within the cell. With the advent of high-throughput techniques, significant amount of protein interaction (PPI) data has been catalogued for organisms such as yeast, which has in turn fueled computational methods for systematic identification and study of protein complexes. However, many complexes are dynamic entities - their subunits are known to assemble at a particular cellular space and time to perform a particular function and disassemble after that - and while current computational analyses have concentrated on studying the dynamics of individual or pairs of proteins in PPI networks, a crucial aspect overlooked is the dynamics of whole complex formations. In this work, using yeast as our model, we incorporate 'time' in the form of cell-cycle phases into the prediction of complexes from PPI networks and study the temporal phenomena of complex assembly and disassembly across phases. We hypothesize that 'staticness' (constitutive expression) of proteins might be related to their temporal "reusability" across complexes, and test this hypothesis using complexes predicted from large-scale PPI networks across the yeast cell cycle phases. Our results hint towards a biological design principle underlying cellular mechanisms - cells maintain generic proteins as 'static' to enable their "reusability" across multiple temporal complexes. We also demonstrate that these findings provide additional support and alternative explanations to findings from existing works on the dynamics in PPI networks.
蛋白质相互作用复合物是驱动细胞内关键生物机制的基本功能单元之一。随着高通量技术的出现,已经为酵母等生物体编目了大量的蛋白质相互作用 (PPI) 数据,这反过来又推动了用于系统识别和研究蛋白质复合物的计算方法的发展。然而,许多复合物是动态实体——它们的亚基已知会在特定的细胞空间和时间组装起来执行特定的功能,然后再分解——虽然当前的计算分析集中在研究 PPI 网络中单个或成对蛋白质的动力学,但一个被忽视的关键方面是整个复合物形成的动力学。在这项工作中,我们使用酵母作为模型,将“时间”以细胞周期阶段的形式纳入从 PPI 网络中预测复合物的过程中,并研究跨阶段复合物组装和分解的时间现象。我们假设蛋白质的“静态性”(组成型表达)可能与其在整个复合物中的时间“可重用性”有关,并使用跨越酵母细胞周期阶段的大规模 PPI 网络预测的复合物来检验这一假设。我们的结果暗示了细胞机制背后存在一种生物学设计原则——细胞将通用蛋白质保持为“静态”,以使其在多个时间复合物中具有“可重用性”。我们还证明,这些发现为 PPI 网络中动力学的现有研究结果提供了额外的支持和替代解释。