Division of Cardiology, Department of Medicine, David Geffen School of Medicine, University of California Los Angeles, CA, USA ; Department of Microbiology, Immunology and Molecular Genetics, University of California Los Angeles, CA, USA.
Front Genet. 2013 Mar 12;4:28. doi: 10.3389/fgene.2013.00028. eCollection 2013.
Network construction and analysis algorithms provide scientists with the ability to sift through high-throughput biological outputs, such as transcription microarrays, for small groups of genes (modules) that are relevant for further research. Most of these algorithms ignore the important role of non-linear interactions in the data, and the ability for genes to operate in multiple functional groups at once, despite clear evidence for both of these phenomena in observed biological systems.
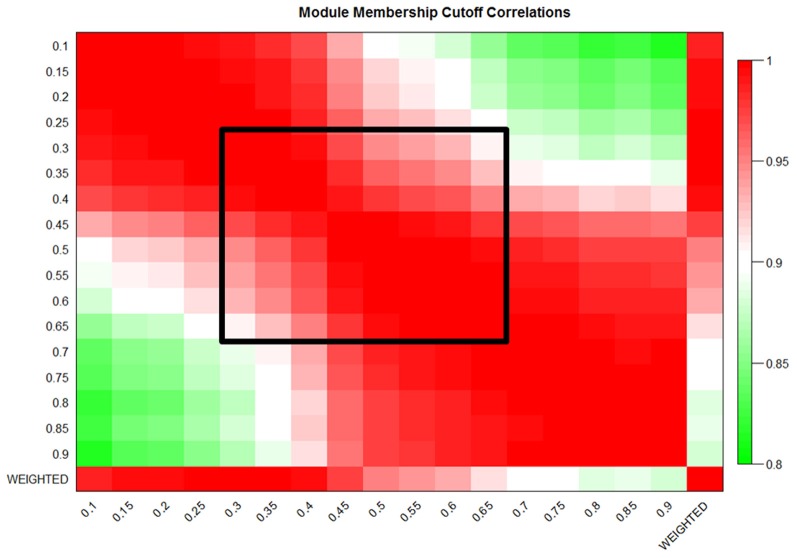
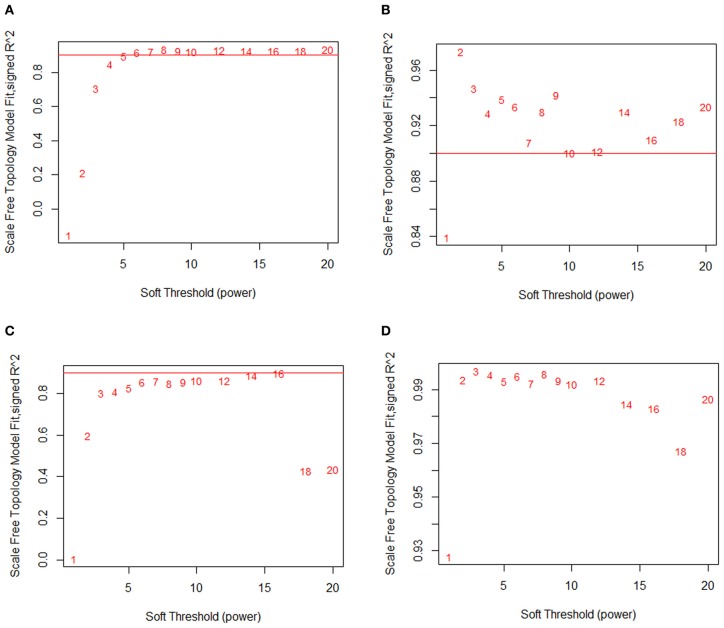
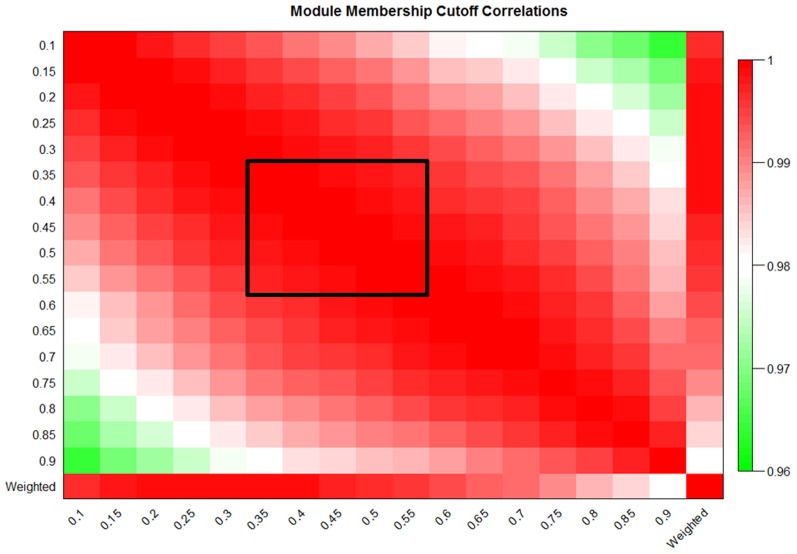
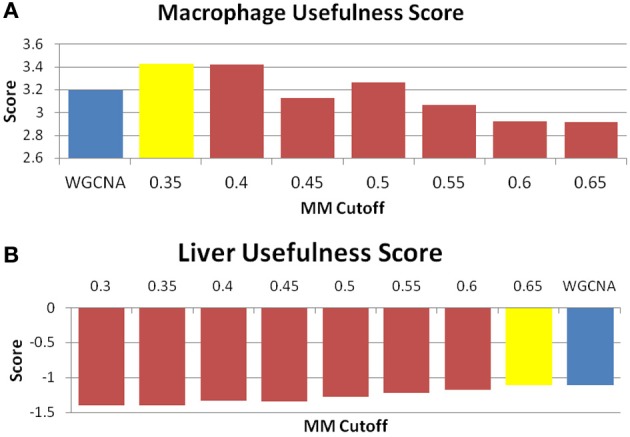
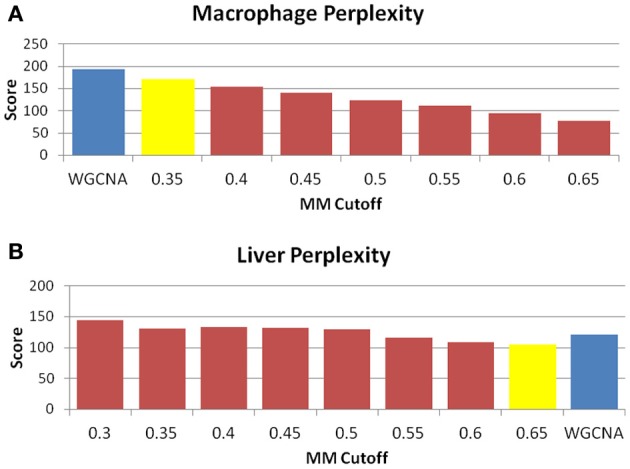
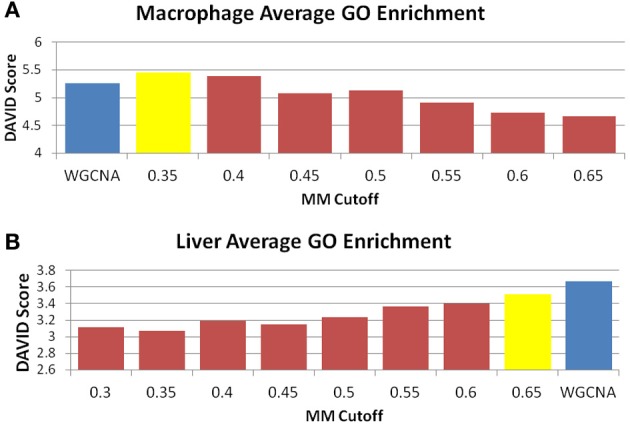
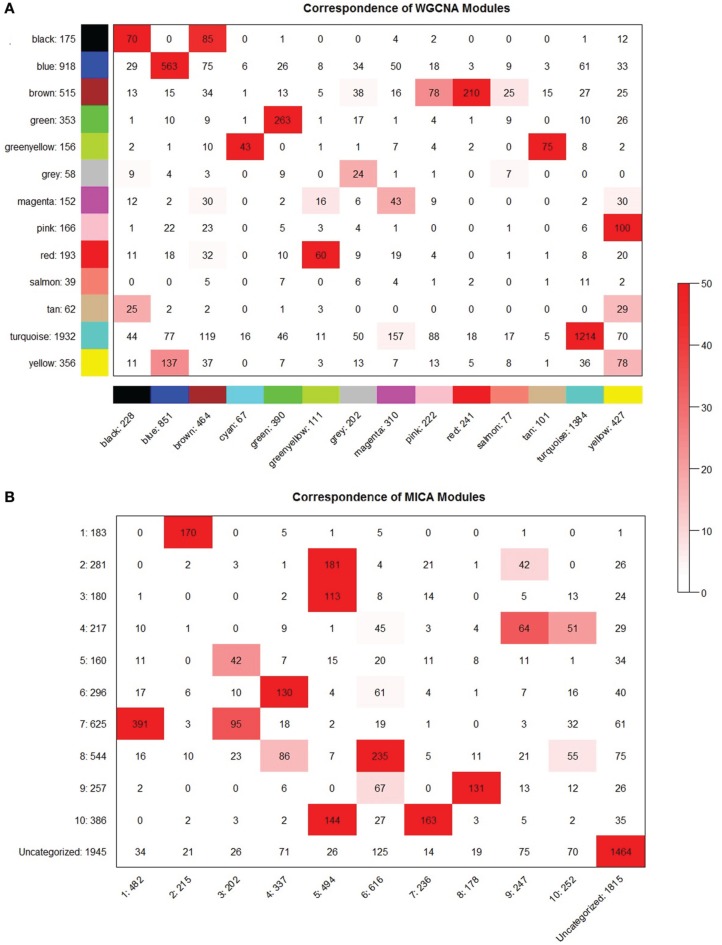
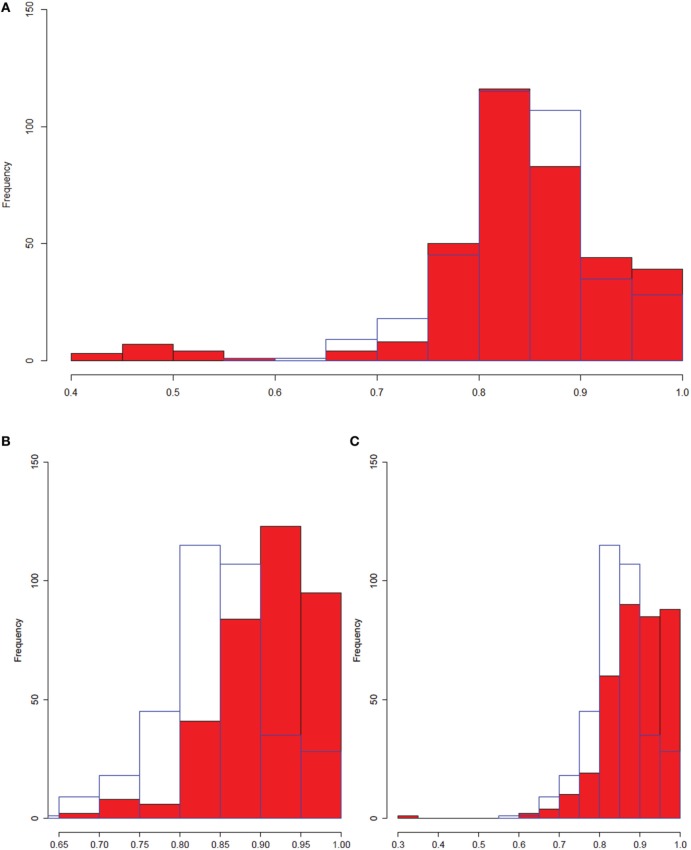
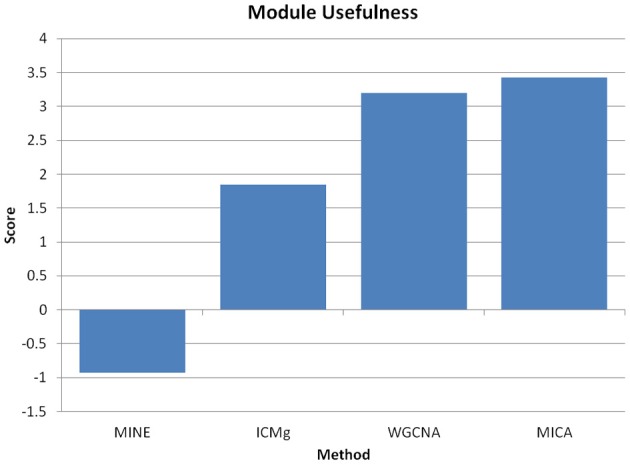
We have created a novel co-expression network analysis algorithm that incorporates both of these principles by combining the information-theoretic association measure of the maximal information coefficient (MIC) with an Interaction Component Model. We evaluate the performance of this approach on two datasets collected from a large panel of mice, one from macrophages and the other from liver by comparing the two measures based on a measure of module entropy, Gene Ontology (GO) enrichment, and scale-free topology (SFT) fit. Our algorithm outperforms a widely used co-expression analysis method, weighted gene co-expression network analysis (WGCNA), in the macrophage data, while returning comparable results in the liver dataset when using these criteria. We demonstrate that the macrophage data has more non-linear interactions than the liver dataset, which may explain the increased performance of our method, termed Maximal Information Component Analysis (MICA) in that case.
In making our network algorithm more accurately reflect known biological principles, we are able to generate modules with improved relevance, particularly in networks with confounding factors such as gene by environment interactions.
网络构建和分析算法为科学家提供了一种能力,可从转录微阵列等高通量生物输出中筛选出与进一步研究相关的小基因群(模块)。这些算法大多数忽略了数据中非线性相互作用的重要作用,以及基因同时在多个功能组中运作的能力,尽管在观察到的生物系统中都有明确的证据表明存在这两种现象。
我们创建了一种新的共表达网络分析算法,通过将最大信息系数(MIC)的信息论关联度量与交互成分模型相结合,结合了这两个原理。我们通过模块熵、基因本体论(GO)富集和无标度拓扑(SFT)拟合这两个度量来比较这两种方法,评估了这种方法在从大量小鼠中收集的两个数据集上的性能,一个来自巨噬细胞,另一个来自肝脏。我们的算法在巨噬细胞数据上优于一种广泛使用的共表达分析方法——加权基因共表达网络分析(WGCNA),而在使用这些标准时,在肝脏数据集上返回了相当的结果。我们证明巨噬细胞数据比肝脏数据集具有更多的非线性相互作用,这可能解释了我们的方法——称为最大信息成分分析(MICA)在这种情况下性能提高的原因。
通过使我们的网络算法更准确地反映已知的生物学原理,我们能够生成相关性更好的模块,特别是在存在基因与环境相互作用等混杂因素的网络中。