Department of Chemistry, University of Southern California , SGM 418, 3620 McClintock Avenue, Los Angeles, California 90089, United States.
J Phys Chem B. 2013 Oct 24;117(42):12807-19. doi: 10.1021/jp4020146. Epub 2013 May 30.
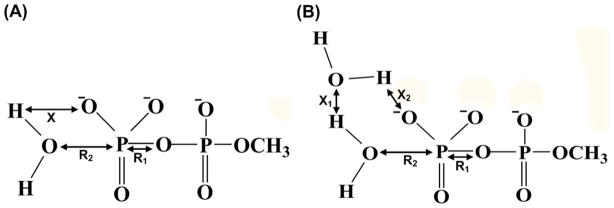
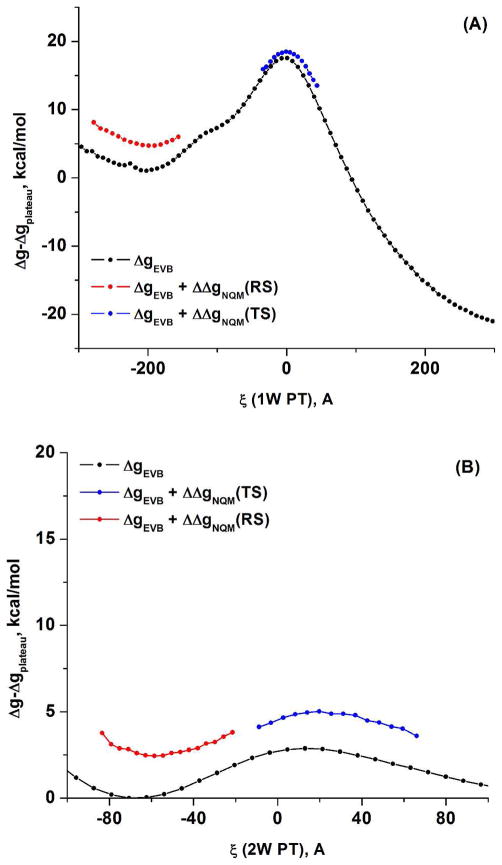
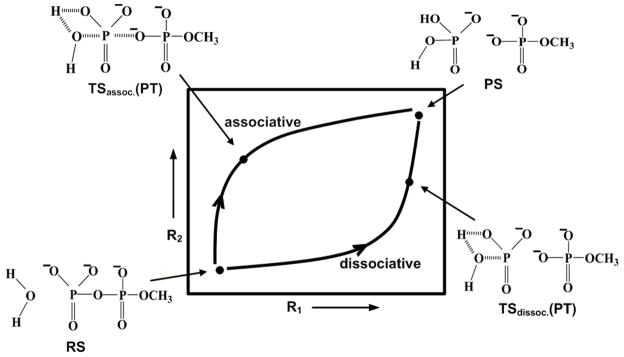
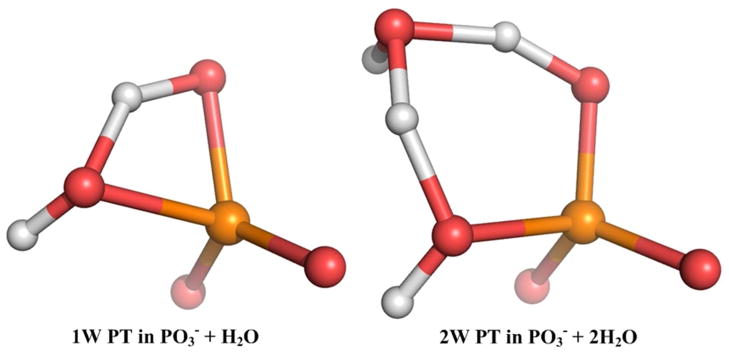
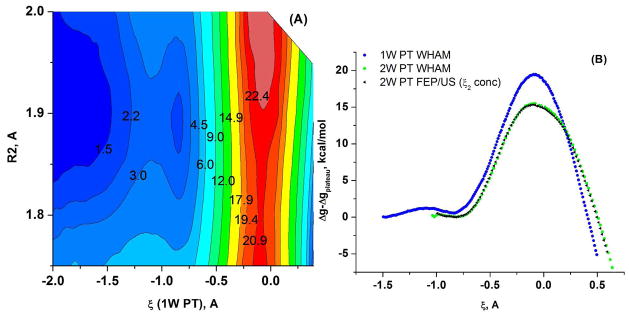
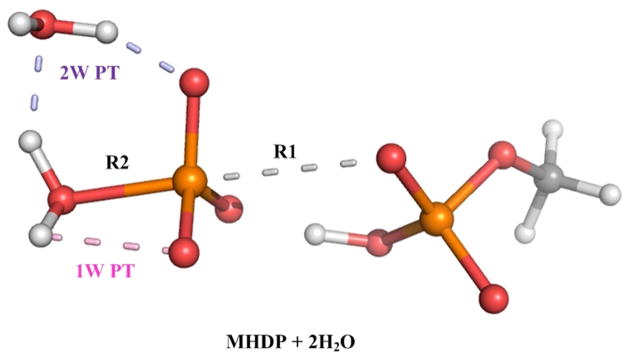
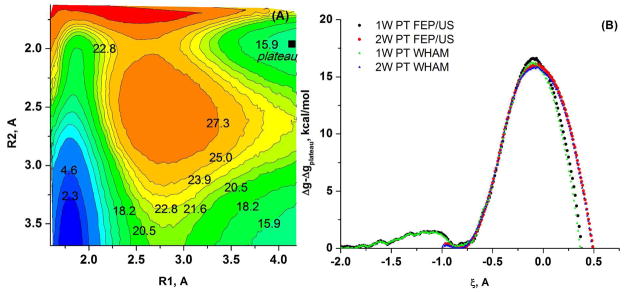
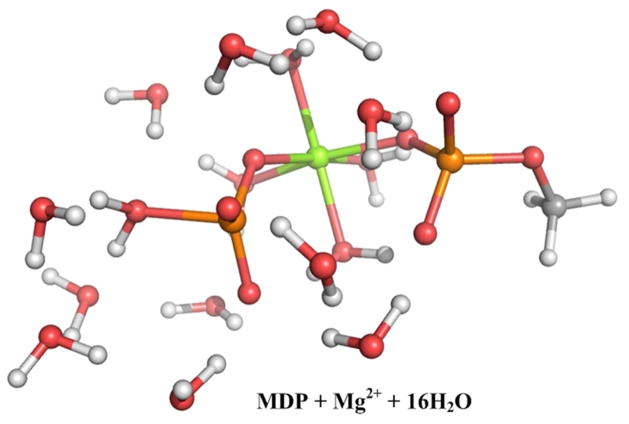
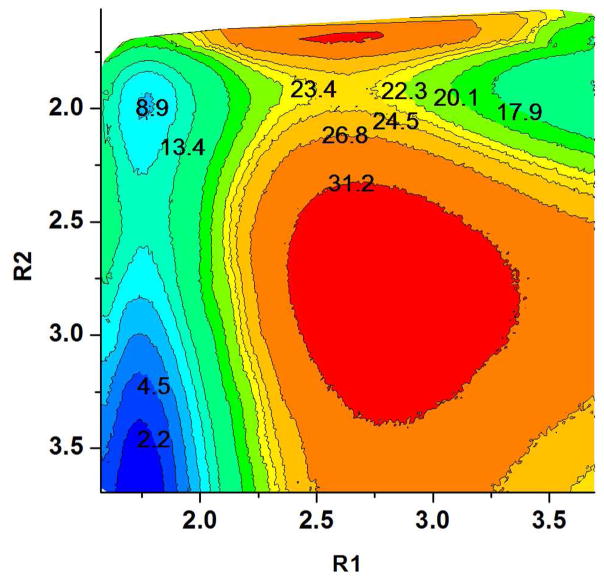
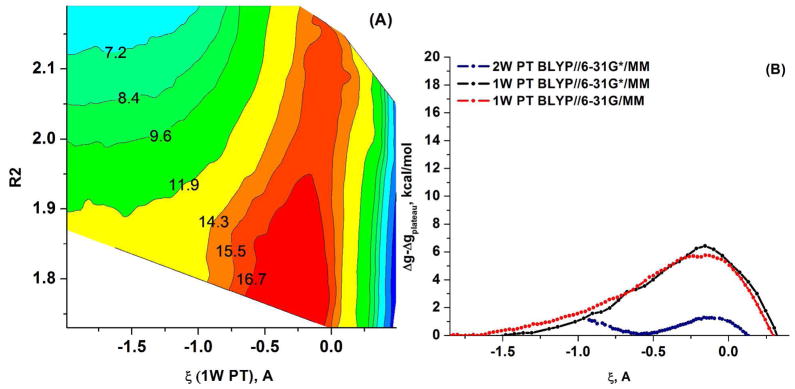
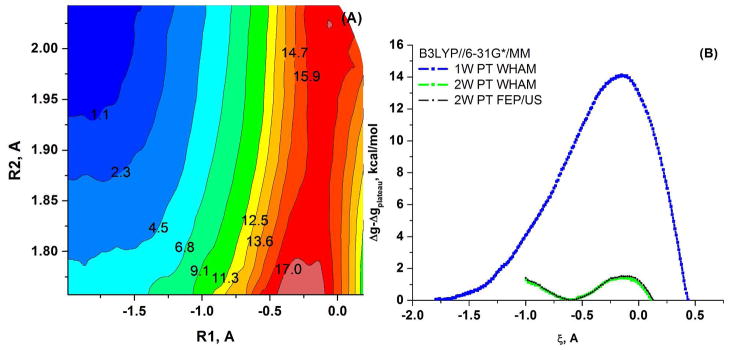
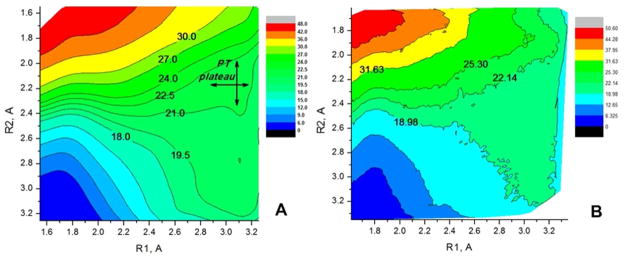
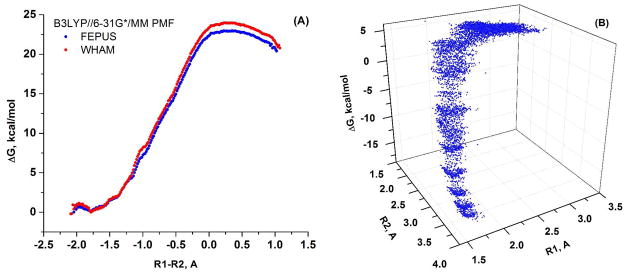
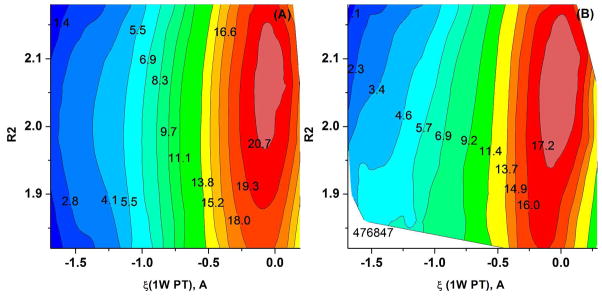
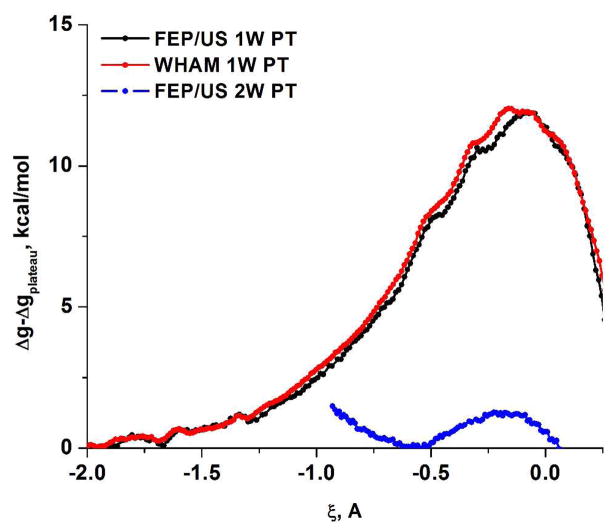
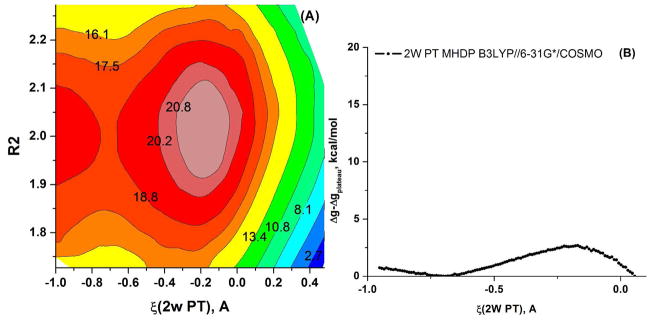
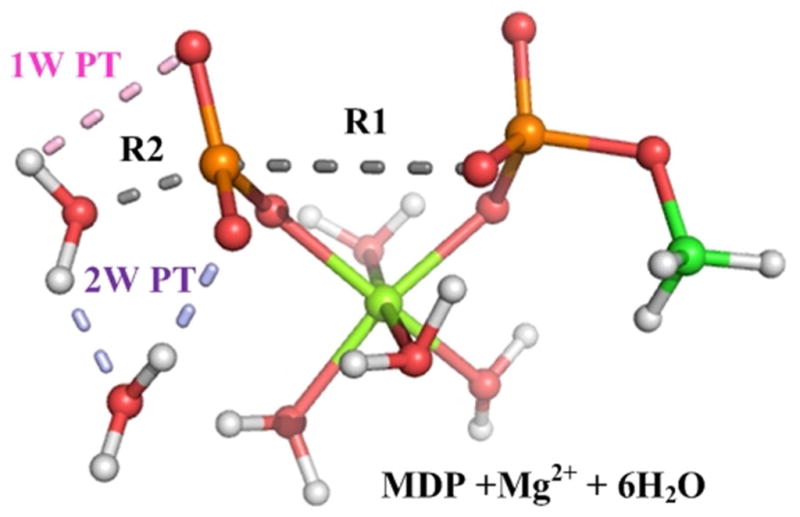
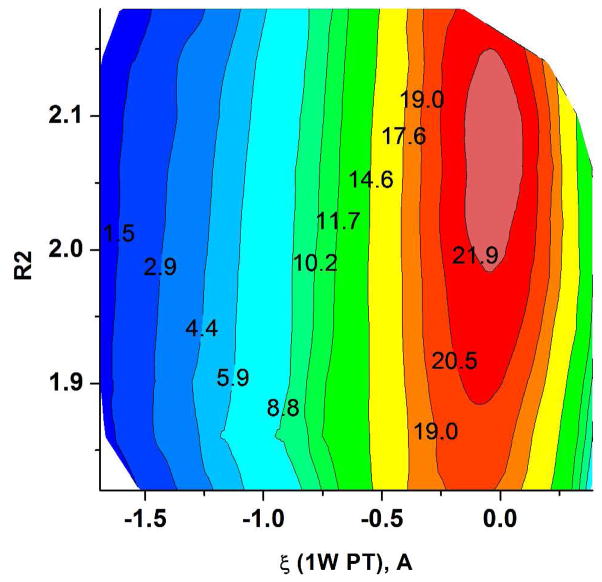
Understanding the nature of the free-energy surfaces for phosphate hydrolysis is a prerequisite for understanding the corresponding key chemical reactions in biology. Here, the challenge has been to move to careful ab initio QM/MM (QM(ai)/MM) free-energy calculations, where obtaining converging results is very demanding and computationally expensive. This work describes such calculations, focusing on the free-energy surface for the hydrolysis of phosphate monoesters, paying special attention to the comparison between the one water (1W) and two water (2W) paths for the proton-transfer (PT) step. This issue has been explored before by energy minimization with implicit solvent models and by nonsystematic QM/MM energy minimization, as well as by nonsystematic free-energy mapping. However, no study has provided the needed reliable 2D (3D) surfaces that are necessary for reaching concrete conclusions. Here we report a systematic evaluation of the 2D (3D) free-energy maps for several relevant systems, comparing the results of QM(ai)/MM and QM(ai)/implicit solvent surfaces, and provide an advanced description of the relevant energetics. It is found that the 1W path for the hydrolysis of the methyl diphosphate (MDP) trianion is 6-9 kcal/mol higher than that the 2W path. This difference becomes slightly larger in the presence of the Mg(2+) ion because this ion reduces the pKa of the conjugated acid form of the phosphate oxygen that accepts the proton. Interestingly, the BLYP approach (which has been used extensively in some studies) gives a much smaller difference between the 1W and 2W activation barriers. At any rate, it is worth pointing out that the 2W transition state for the PT is not much higher that the common plateau that serves as the starting point of both the 1W and 2W PT paths. Thus, the calculated catalytic effects of proteins based on the 2W PT mechanistic model are not expected to be different from the catalytic effects predicted using the 1W PT mechanistic model, which was calibrated on the observed barrier in solution and in which the TS charge distribution was similar to the that of the plateau (as was done in all of our previous EVB studies).
理解磷酸盐水解的自由能表面的性质是理解生物学中相应关键化学反应的前提。在这里,挑战在于转向仔细的从头算 QM/MM(QM(ai)/MM)自由能计算,其中获得收敛的结果是非常苛刻和昂贵的。这项工作描述了这样的计算,重点是磷酸盐单酯水解的自由能表面,特别关注质子转移(PT)步骤中一个水分子(1W)和两个水分子(2W)路径之间的比较。这个问题以前已经通过使用隐式溶剂模型的能量最小化以及非系统的 QM/MM 能量最小化,以及非系统的自由能映射进行了探讨。然而,没有研究提供了为得出具体结论所需的可靠的 2D(3D)表面。在这里,我们报告了对几个相关系统的 2D(3D)自由能图的系统评估,比较了 QM(ai)/MM 和 QM(ai)/隐式溶剂表面的结果,并提供了相关能量学的高级描述。结果发现,MDP 三阴离子水解的 1W 路径比 2W 路径高 6-9 kcal/mol。在存在 Mg(2+)离子的情况下,这种差异会略大一些,因为该离子降低了接受质子的磷酸氧的共轭酸形式的 pKa。有趣的是,BLYP 方法(在一些研究中被广泛使用)在 1W 和 2W 活化能垒之间给出了更小的差异。无论如何,值得指出的是,PT 的 2W 过渡态并不比作为 1W 和 2W PT 途径起点的常见平台高很多。因此,基于 2W PT 机制模型的蛋白质的计算催化效应不应与使用在溶液中观察到的屏障校准的 1W PT 机制模型预测的催化效应不同,在该模型中,TS 电荷分布与平台相似(在我们之前的所有 EVB 研究中都是如此)。