Bioinformatic Centre, Institute of Microbial Technology, Chandigarh, India.
PLoS One. 2013 Jun 28;8(6):e67008. doi: 10.1371/journal.pone.0067008. Print 2013.
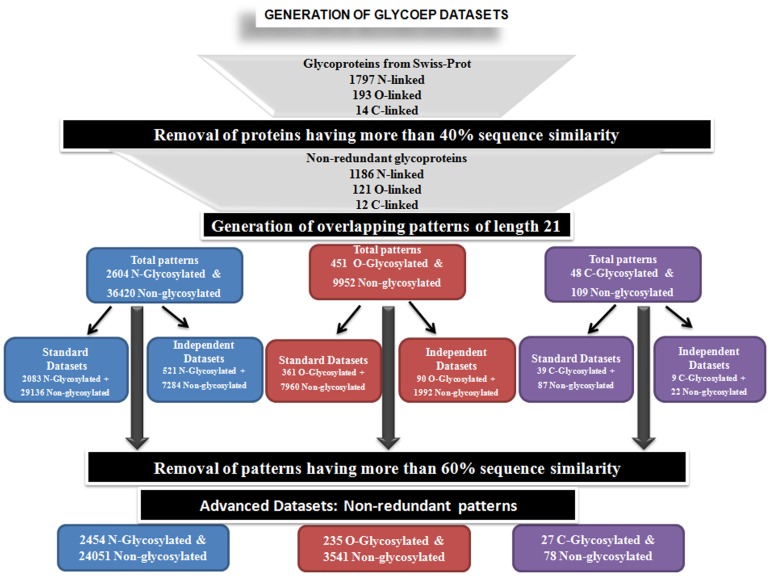
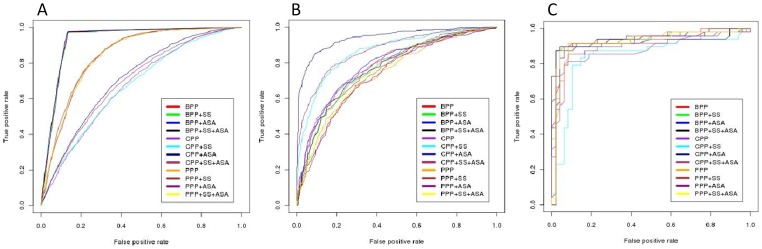
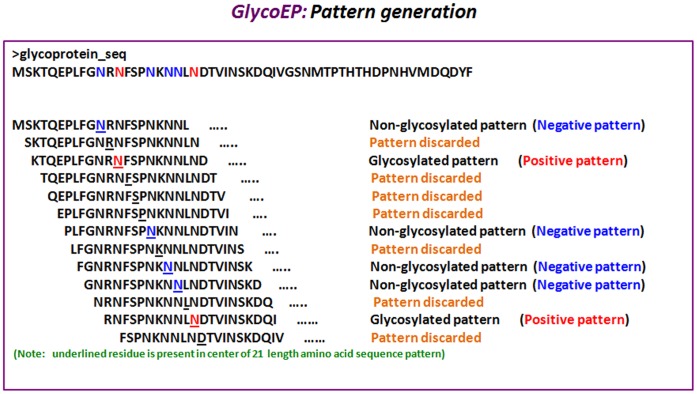
Glycosylation is one of the most abundant and an important post-translational modification of proteins. Glycosylated proteins (glycoproteins) are involved in various cellular biological functions like protein folding, cell-cell interactions, cell recognition and host-pathogen interactions. A large number of eukaryotic glycoproteins also have therapeutic and potential technology applications. Therefore, characterization and analysis of glycosites (glycosylated residues) in these proteins is of great interest to biologists. In order to cater these needs a number of in silico tools have been developed over the years, however, a need to get even better prediction tools remains. Therefore, in this study we have developed a new webserver GlycoEP for more accurate prediction of N-linked, O-linked and C-linked glycosites in eukaryotic glycoproteins using two larger datasets, namely, standard and advanced datasets. In case of standard datasets no two glycosylated proteins are more similar than 40%; advanced datasets are highly non-redundant where no two glycosites' patterns (as defined in methods) have more than 60% similarity. Further, based on our results with several algorihtms developed using different machine-learning techniques, we found Support Vector Machine (SVM) as optimum tool to develop glycosite prediction models. Accordingly, using our more stringent and non-redundant advanced datasets, the SVM based models developed in this study achieved a prediction accuracy of 84.26%, 86.87% and 91.43% with corresponding MCC of 0.54, 0.20 and 0.78, for N-, O- and C-linked glycosites, respectively. The best performing models trained on advanced datasets were then implemented as a user-friendly web server GlycoEP (http://www.imtech.res.in/raghava/glycoep/). Additionally, this server provides prediction models developed on standard datasets and allows users to scan sequons in input protein sequences.
糖基化是蛋白质最丰富和最重要的翻译后修饰之一。糖基化蛋白(glycoproteins)参与各种细胞生物学功能,如蛋白质折叠、细胞-细胞相互作用、细胞识别和宿主-病原体相互作用。大量真核糖蛋白也具有治疗和潜在的技术应用。因此,对生物学家来说,对这些蛋白质中的糖基化位点(glycosylated residues)进行特征描述和分析非常重要。为了满足这些需求,多年来已经开发了许多计算机工具,但是仍然需要更好的预测工具。因此,在这项研究中,我们使用两个更大的数据集(标准数据集和高级数据集)开发了一种新的网络服务器 GlycoEP,用于更准确地预测真核糖蛋白中的 N-连接、O-连接和 C-连接糖基化位点。对于标准数据集,没有两个糖基化蛋白彼此相似超过 40%;高级数据集高度非冗余,其中没有两个糖基化位点模式(如方法中定义)具有超过 60%的相似性。此外,根据我们使用不同机器学习技术开发的几种算法的结果,我们发现支持向量机(SVM)是开发糖基化预测模型的最佳工具。因此,使用我们更严格和非冗余的高级数据集,本研究中开发的基于 SVM 的模型在预测 N-、O-和 C-连接糖基化位点时,分别达到了 84.26%、86.87%和 91.43%的准确率,相应的 MCC 为 0.54、0.20 和 0.78。然后,将在高级数据集上训练的表现最佳的模型实现为一个用户友好的网络服务器 GlycoEP(http://www.imtech.res.in/raghava/glycoep/)。此外,该服务器还提供基于标准数据集开发的预测模型,并允许用户扫描输入蛋白质序列中的信号肽。