Joint Center for Structural Genomics, Stanford Synchrotron Radiation Lightsource, Stanford, California, USA.
Nat Methods. 2013 Sep;10(9):896-902. doi: 10.1038/nmeth.2592. Epub 2013 Aug 4.
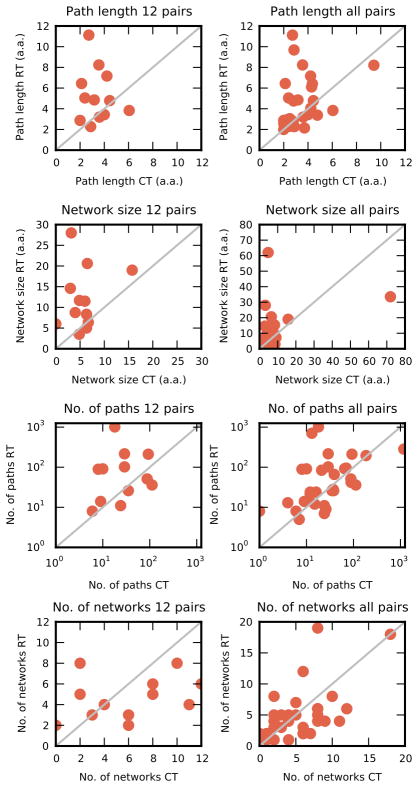
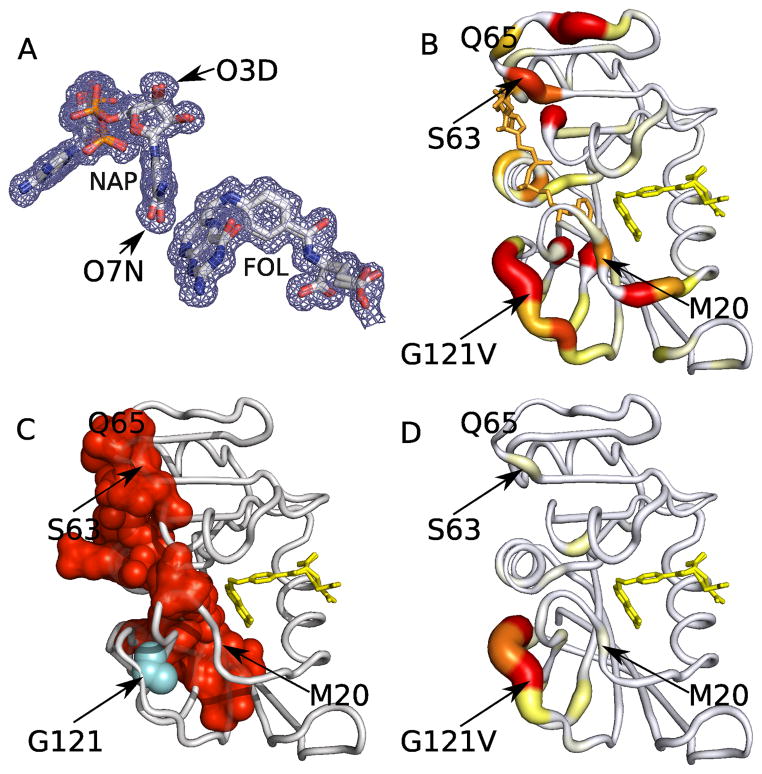
Protein function often depends on the exchange between conformational substates. Allosteric ligand binding or distal mutations can stabilize specific active-site conformations and consequently alter protein function. Observing alternative conformations at low levels of electron density, in addition to comparison of independently determined X-ray crystal structures, can provide mechanistic insights into conformational dynamics. Here we report a new algorithm, CONTACT, that identifies contact networks of conformationally heterogeneous residues directly from high-resolution X-ray crystallography data. Contact networks determined for Escherichia coli dihydrofolate reductase (ecDHFR) predict the observed long-range pattern of NMR chemical shift perturbations of an allosteric mutation. A comparison of contact networks in wild-type and mutant ecDHFR suggests that mutations that alter optimized contact networks of coordinated motions can impair catalytic function. CONTACT-guided mutagenesis can exploit the structure-dynamics-function relationship in protein engineering and design.
蛋白质的功能通常取决于构象亚稳态之间的交换。变构配体结合或远端突变可以稳定特定的活性位点构象,从而改变蛋白质的功能。在低电子密度水平下观察到的替代构象,以及独立确定的 X 射线晶体结构的比较,可以提供对构象动力学的机制见解。在这里,我们报告了一种新的算法 CONTACT,它可以直接从高分辨率 X 射线晶体学数据中识别构象异质残基的接触网络。为大肠杆菌二氢叶酸还原酶(ecDHFR)确定的接触网络预测了变构突变的 NMR 化学位移扰动的观察到的远程模式。野生型和突变型 ecDHFR 中接触网络的比较表明,改变协调运动优化接触网络的突变可能会损害催化功能。CONTACT 指导的诱变可以利用蛋白质工程和设计中的结构-动力学-功能关系。