Department of Cancer Immunotherapeutics and Tumor Immunology, City of Hope and Beckman Research Institute, Duarte, California, United States of America.
PLoS Comput Biol. 2013;9(8):e1003189. doi: 10.1371/journal.pcbi.1003189. Epub 2013 Aug 22.
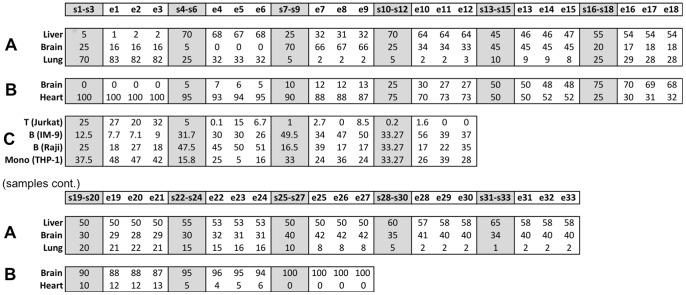
Gene expression analysis is generally performed on heterogeneous tissue samples consisting of multiple cell types. Current methods developed to separate heterogeneous gene expression rely on prior knowledge of the cell-type composition and/or signatures--these are not available in most public datasets. We present a novel method to identify the cell-type composition, signatures and proportions per sample without need for a-priori information. The method was successfully tested on controlled and semi-controlled datasets and performed as accurately as current methods that do require additional information. As such, this method enables the analysis of cell-type specific gene expression using existing large pools of publically available microarray datasets.
基因表达分析通常在由多种细胞类型组成的异质组织样本上进行。目前开发的用于分离异质基因表达的方法依赖于对细胞类型组成和/或特征的先验知识——这些在大多数公共数据集上是不可用的。我们提出了一种新的方法,可以在无需先验信息的情况下识别每个样本的细胞类型组成、特征和比例。该方法在受控和半受控数据集上进行了成功测试,其准确性与需要额外信息的现有方法相当。因此,该方法可以使用现有的大量公共微阵列数据集来分析细胞类型特异性基因表达。