Programa Nacional de Investigación Cultivos de Secano, Instituto Nacional de Investigación Agropecuaria, Est. Exp. La Estanzuela, Colonia 70000, Uruguay.
G3 (Bethesda). 2013 Dec 9;3(12):2105-14. doi: 10.1534/g3.113.007807.
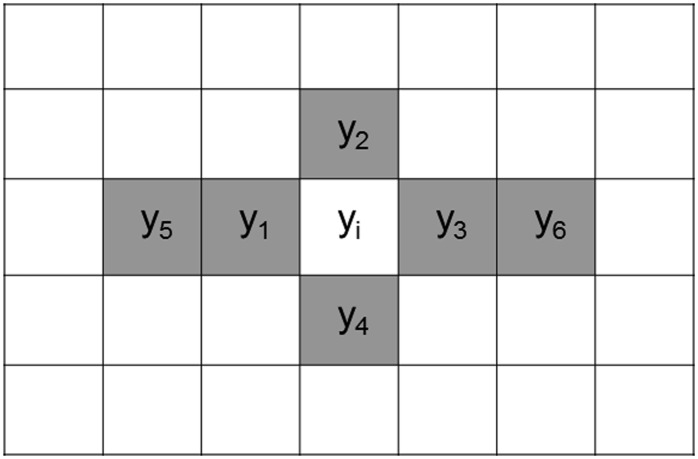
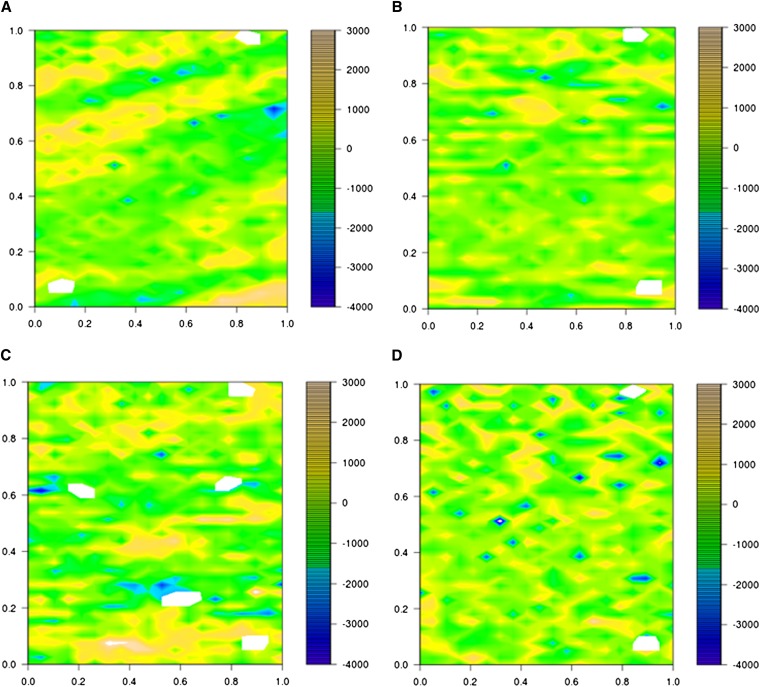
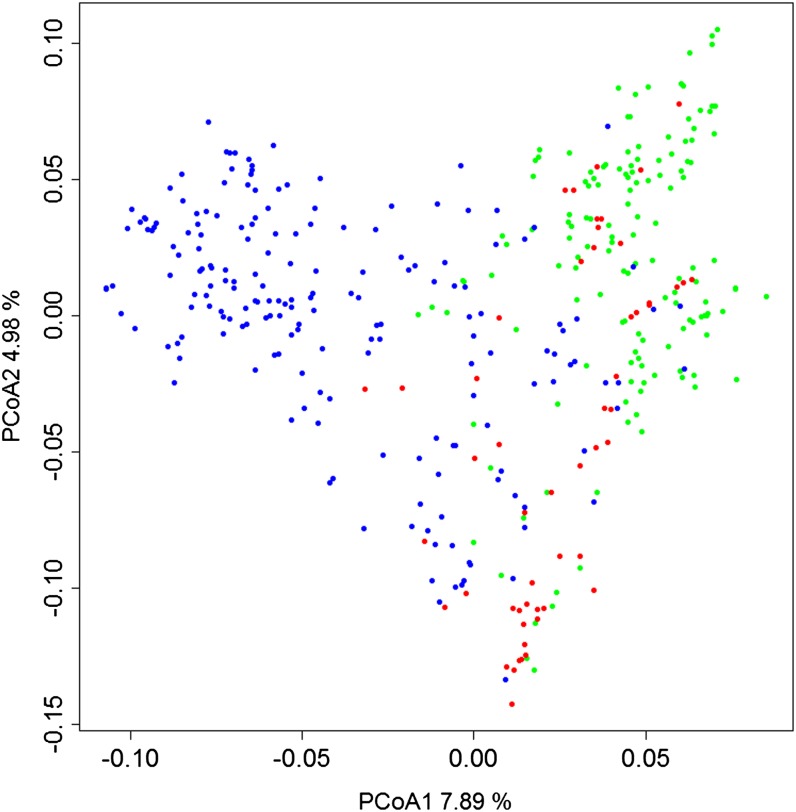
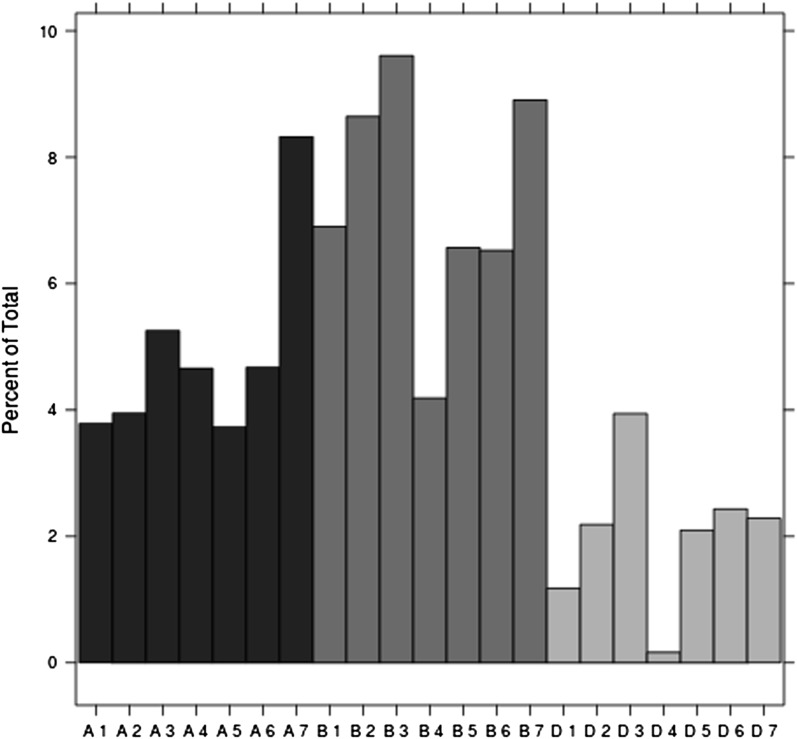
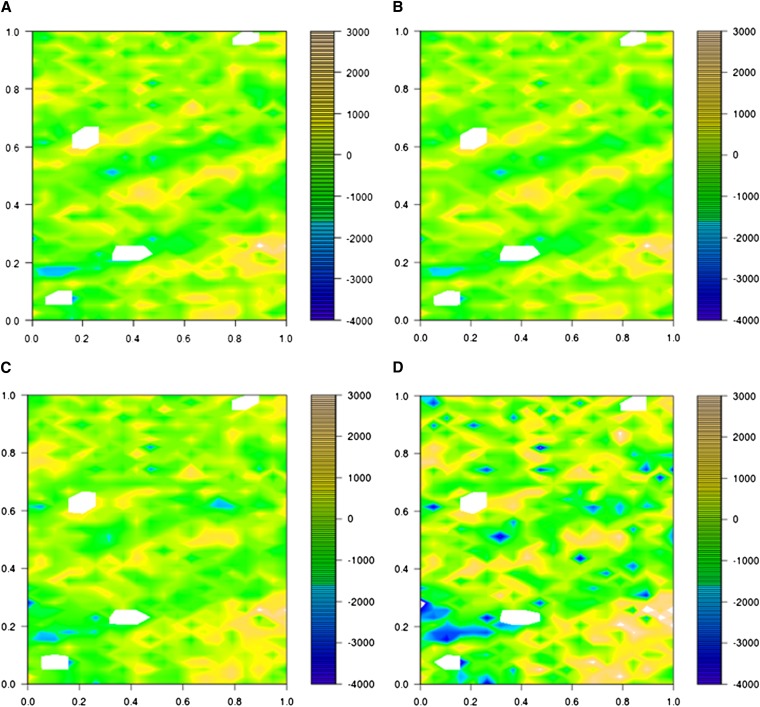
In crop breeding, the interest of predicting the performance of candidate cultivars in the field has increased due to recent advances in molecular breeding technologies. However, the complexity of the wheat genome presents some challenges for applying new technologies in molecular marker identification with next-generation sequencing. We applied genotyping-by-sequencing, a recently developed method to identify single-nucleotide polymorphisms, in the genomes of 384 wheat (Triticum aestivum) genotypes that were field tested under three different water regimes in Mediterranean climatic conditions: rain-fed only, mild water stress, and fully irrigated. We identified 102,324 single-nucleotide polymorphisms in these genotypes, and the phenotypic data were used to train and test genomic selection models intended to predict yield, thousand-kernel weight, number of kernels per spike, and heading date. Phenotypic data showed marked spatial variation. Therefore, different models were tested to correct the trends observed in the field. A mixed-model using moving-means as a covariate was found to best fit the data. When we applied the genomic selection models, the accuracy of predicted traits increased with spatial adjustment. Multiple genomic selection models were tested, and a Gaussian kernel model was determined to give the highest accuracy. The best predictions between environments were obtained when data from different years were used to train the model. Our results confirm that genotyping-by-sequencing is an effective tool to obtain genome-wide information for crops with complex genomes, that these data are efficient for predicting traits, and that correction of spatial variation is a crucial ingredient to increase prediction accuracy in genomic selection models.
在作物育种中,由于分子育种技术的最新进展,预测候选品种在田间表现的兴趣有所增加。然而,小麦基因组的复杂性为应用新技术在分子标记识别方面带来了一些挑战,这些新技术采用的是下一代测序。我们应用了全基因组重测序技术(genotyping-by-sequencing),这是一种最近开发的用于识别单核苷酸多态性的方法,对 384 个小麦(Triticum aestivum)基因型的基因组进行了分析,这些基因型在三种不同的水条件下进行了田间试验,包括仅降雨、轻度水分胁迫和充分灌溉。我们在这些基因型中鉴定出了 102324 个单核苷酸多态性,并且使用表型数据来训练和测试旨在预测产量、千粒重、每穗粒数和抽穗期的基因组选择模型。表型数据显示出明显的空间变化。因此,测试了不同的模型来纠正田间观察到的趋势。结果发现,使用移动平均值作为协变量的混合模型最适合拟合数据。当我们应用基因组选择模型时,预测性状的准确性随着空间调整而增加。测试了多个基因组选择模型,发现高斯核模型的准确性最高。当使用不同年份的数据来训练模型时,在不同环境之间获得了最佳的预测结果。我们的结果证实,全基因组重测序是一种获得复杂基因组作物全基因组信息的有效工具,这些数据对预测性状有效,并且纠正空间变化是提高基因组选择模型预测准确性的关键因素。