1] Department of Genetics, Stanford University School of Medicine, Stanford, California, USA. [2] Howard Hughes Medical Institute, Stanford University School of Medicine, Stanford, California, USA. [3] Program in Epithelial Biology, Stanford University School of Medicine, Stanford, California, USA.
Nat Methods. 2013 Dec;10(12):1213-8. doi: 10.1038/nmeth.2688. Epub 2013 Oct 6.
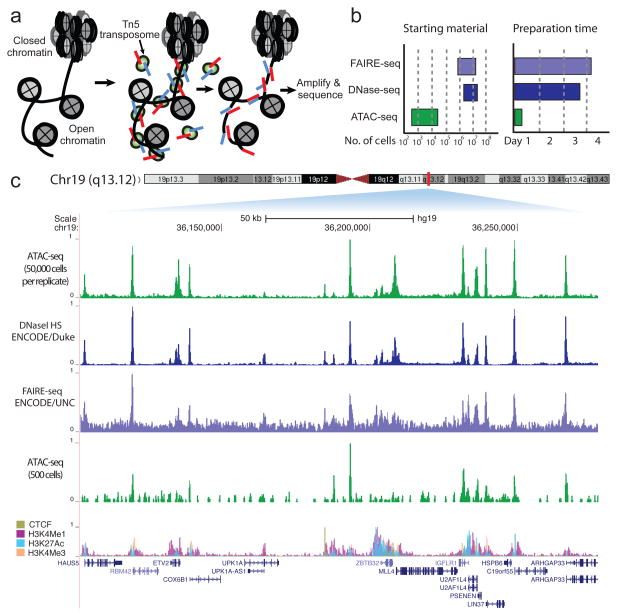
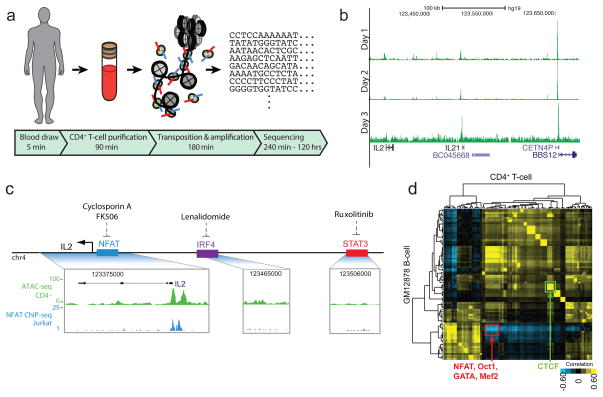
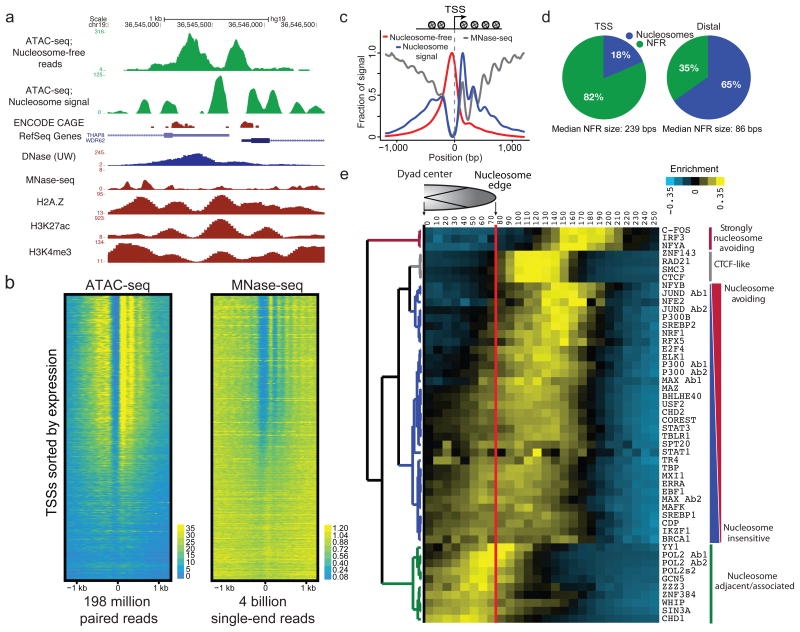
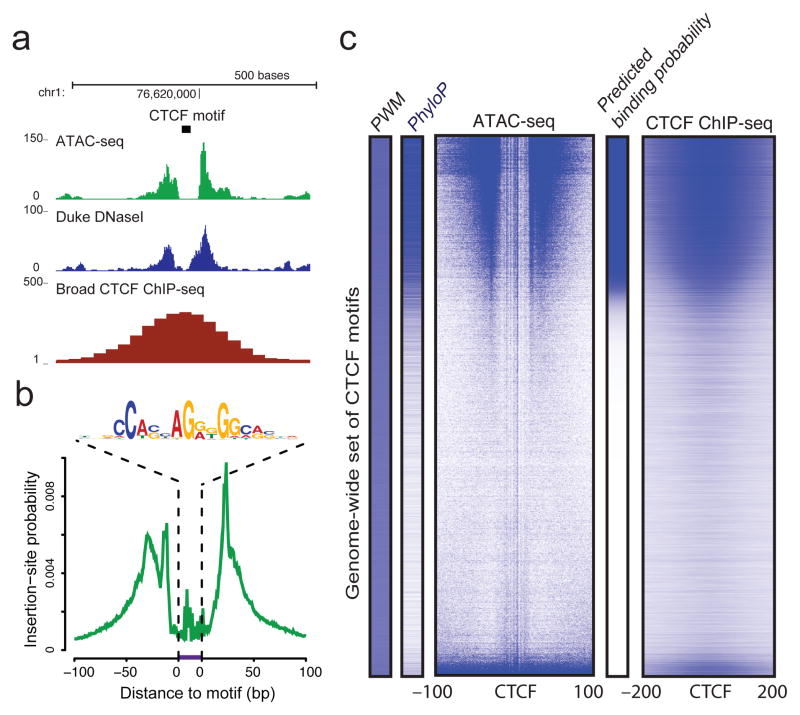
We describe an assay for transposase-accessible chromatin using sequencing (ATAC-seq), based on direct in vitro transposition of sequencing adaptors into native chromatin, as a rapid and sensitive method for integrative epigenomic analysis. ATAC-seq captures open chromatin sites using a simple two-step protocol with 500-50,000 cells and reveals the interplay between genomic locations of open chromatin, DNA-binding proteins, individual nucleosomes and chromatin compaction at nucleotide resolution. We discovered classes of DNA-binding factors that strictly avoided, could tolerate or tended to overlap with nucleosomes. Using ATAC-seq maps of human CD4(+) T cells from a proband obtained on consecutive days, we demonstrated the feasibility of analyzing an individual's epigenome on a timescale compatible with clinical decision-making.
我们描述了一种使用测序进行转座酶可及染色质分析(ATAC-seq)的方法,该方法基于将测序接头直接体外转座到天然染色质中,是一种快速、灵敏的整合表观基因组分析方法。ATAC-seq 使用简单的两步法,用 500-50,000 个细胞捕获开放染色质位点,并在核苷酸分辨率上揭示了开放染色质、DNA 结合蛋白、单个核小体和染色质紧缩之间的相互作用。我们发现了一些 DNA 结合因子的类别,它们严格回避、可以容忍或倾向于与核小体重叠。使用从连续几天获得的供体的人 CD4(+) T 细胞的 ATAC-seq 图谱,我们证明了在与临床决策制定兼容的时间尺度上分析个体表观基因组的可行性。