Gill Andrew C
The Roslin Institute and Royal (Dick) School of Veterinary Studies, Easter Bush Campus, University of Edinburgh, Roslin, Edinburgh, United Kingdom.
PLoS One. 2014 Jan 31;9(1):e87354. doi: 10.1371/journal.pone.0087354. eCollection 2014.
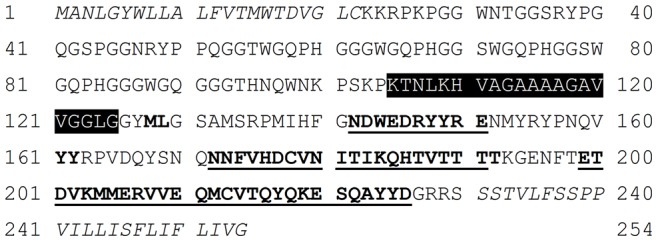
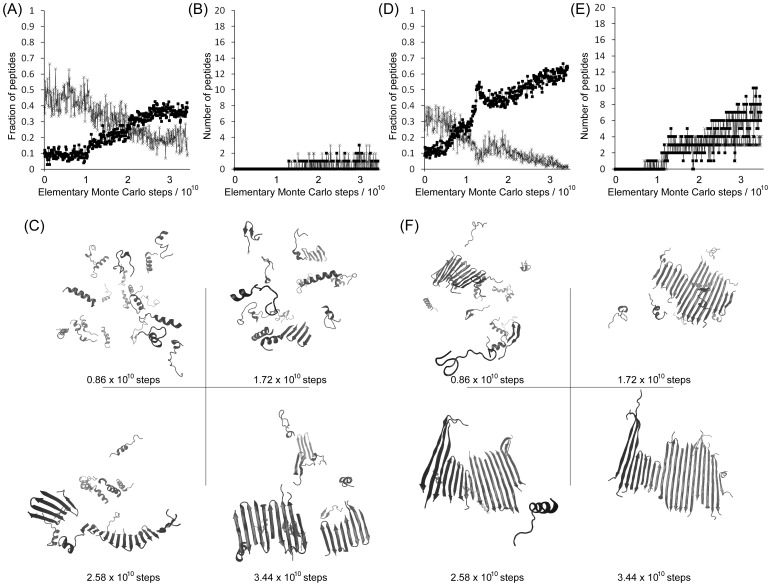
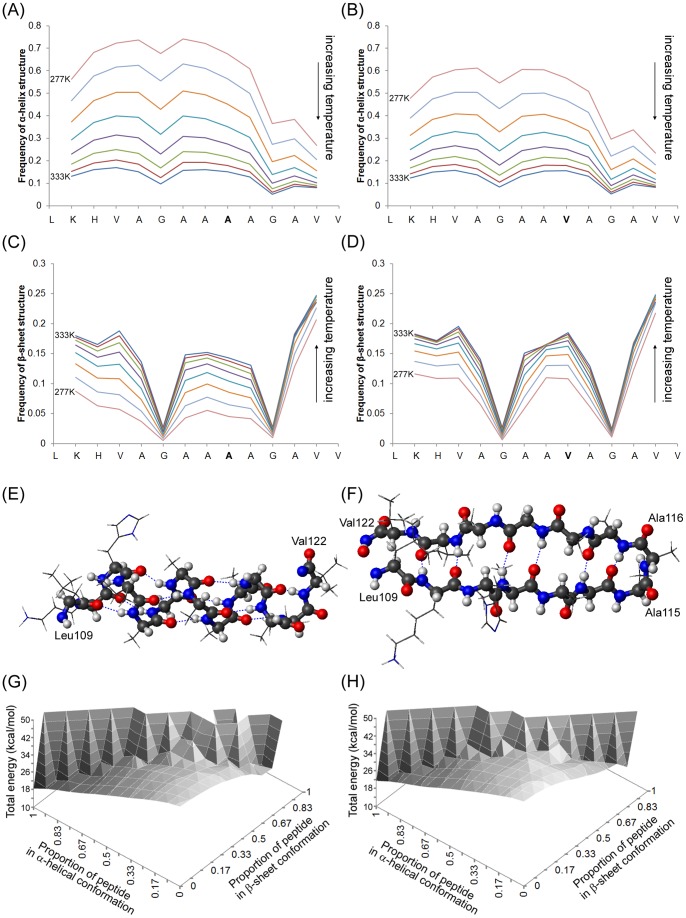
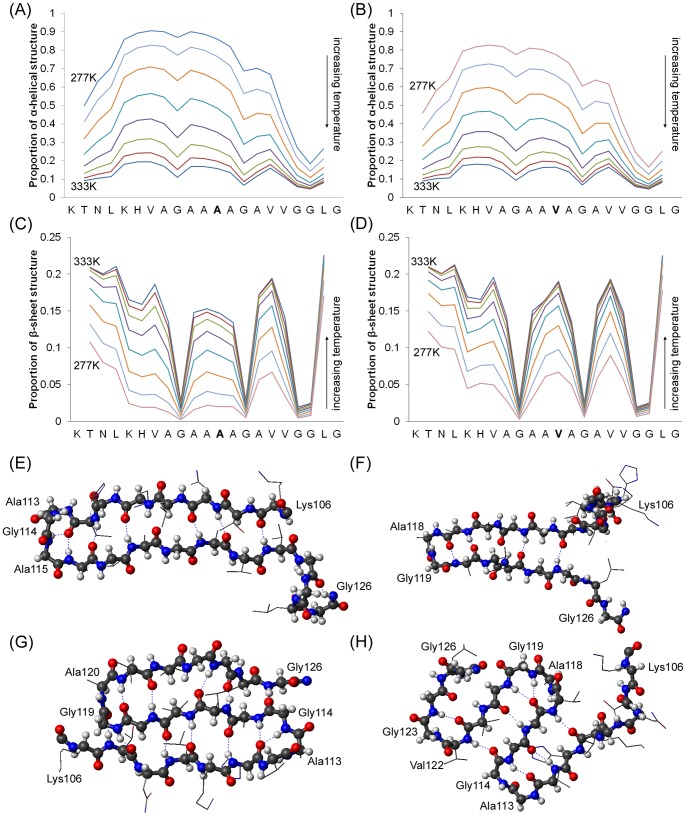
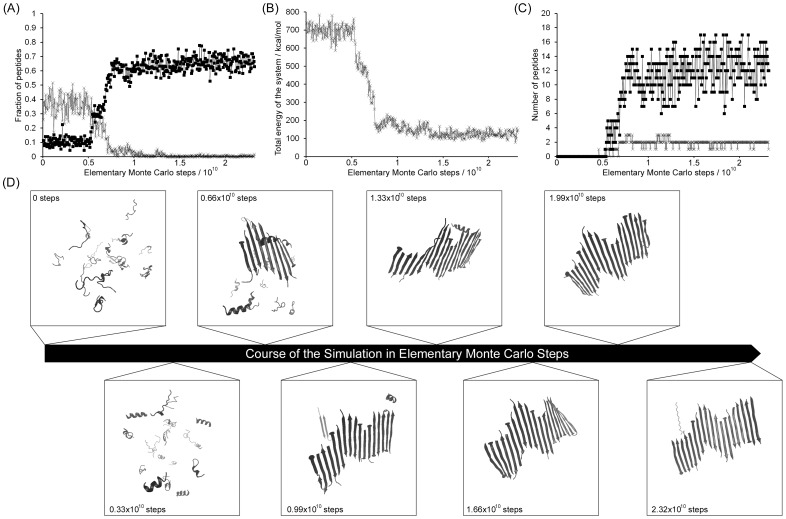
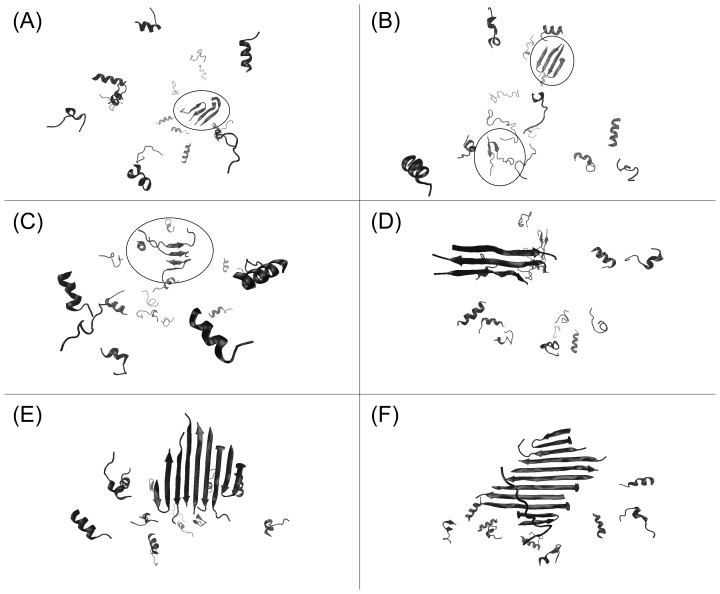
Protein misfolding disorders are associated with conformational changes in specific proteins, leading to the formation of potentially neurotoxic amyloid fibrils. During pathogenesis of prion disease, the prion protein misfolds into β-sheet rich, protease-resistant isoforms. A key, hydrophobic domain within the prion protein, comprising residues 109-122, recapitulates many properties of the full protein, such as helix-to-sheet structural transition, formation of fibrils and cytotoxicity of the misfolded isoform. Using all-atom, molecular simulations, it is demonstrated that the monomeric 109-122 peptide has a preference for α-helical conformations, but that this peptide can also form β-hairpin structures resulting from turns around specific glycine residues of the peptide. Altering a single amino acid within the 109-122 peptide (A117V, associated with familial prion disease) increases the prevalence of β-hairpin formation and these observations are replicated in a longer peptide, comprising residues 106-126. Multi-molecule simulations of aggregation yield different assemblies of peptide molecules composed of conformationally-distinct monomer units. Small molecular assemblies, consistent with oligomers, comprise peptide monomers in a β-hairpin-like conformation and in many simulations appear to exist only transiently. Conversely, larger assemblies are comprised of extended peptides in predominately antiparallel β-sheets and are stable relative to the length of the simulations. These larger assemblies are consistent with amyloid fibrils, show cross-β structure and can form through elongation of monomer units within pre-existing oligomers. In some simulations, assemblies containing both β-hairpin and linear peptides are evident. Thus, in this work oligomers are on pathway to fibril formation and a preference for β-hairpin structure should enhance oligomer formation whilst inhibiting maturation into fibrils. These simulations provide an important new atomic-level model for the formation of oligomers and fibrils of the prion protein and suggest that stabilization of β-hairpin structure may enhance cellular toxicity by altering the balance between oligomeric and fibrillar protein assemblies.
蛋白质错误折叠疾病与特定蛋白质的构象变化有关,导致潜在神经毒性淀粉样原纤维的形成。在朊病毒疾病的发病过程中,朊病毒蛋白错误折叠成富含β-折叠、抗蛋白酶的异构体。朊病毒蛋白内一个关键的疏水结构域,包含第109 - 122位氨基酸残基,概括了完整蛋白质的许多特性,如从螺旋到折叠的结构转变、原纤维的形成以及错误折叠异构体的细胞毒性。通过全原子分子模拟表明,单体的109 - 122肽段倾向于α-螺旋构象,但该肽段也可形成由肽段特定甘氨酸残基处的转角产生的β-发夹结构。改变109 - 122肽段内的单个氨基酸(A117V,与家族性朊病毒疾病相关)会增加β-发夹形成的发生率,并且这些观察结果在包含第106 - 126位氨基酸残基的更长肽段中得到重现。聚集的多分子模拟产生了由构象不同的单体单元组成的肽分子的不同组装形式。与寡聚体一致的小分子组装体,由呈β-发夹样构象的肽单体组成,并且在许多模拟中似乎仅短暂存在。相反,较大的组装体由主要呈反平行β-折叠的延伸肽组成,并且相对于模拟时长是稳定的。这些较大的组装体与淀粉样原纤维一致,显示出交叉β结构,并且可以通过预先存在的寡聚体内单体单元的延伸形成。在一些模拟中,同时含有β-发夹和线性肽的组装体很明显。因此,在这项工作中,寡聚体处于原纤维形成的途径上,并且对β-发夹结构的偏好应会增强寡聚体的形成,同时抑制其成熟为原纤维。这些模拟为朊病毒蛋白寡聚体和原纤维的形成提供了一个重要的新的原子水平模型,并表明β-发夹结构的稳定可能通过改变寡聚体和纤维状蛋白组装体之间的平衡来增强细胞毒性。