Várnai Csilla, Burkoff Nikolas S, Wild David L
Systems Biology Centre, University of Warwick , Coventry, United Kingdom.
J Chem Theory Comput. 2013 Dec 10;9(12):5718-5733. doi: 10.1021/ct400628h. Epub 2013 Nov 15.
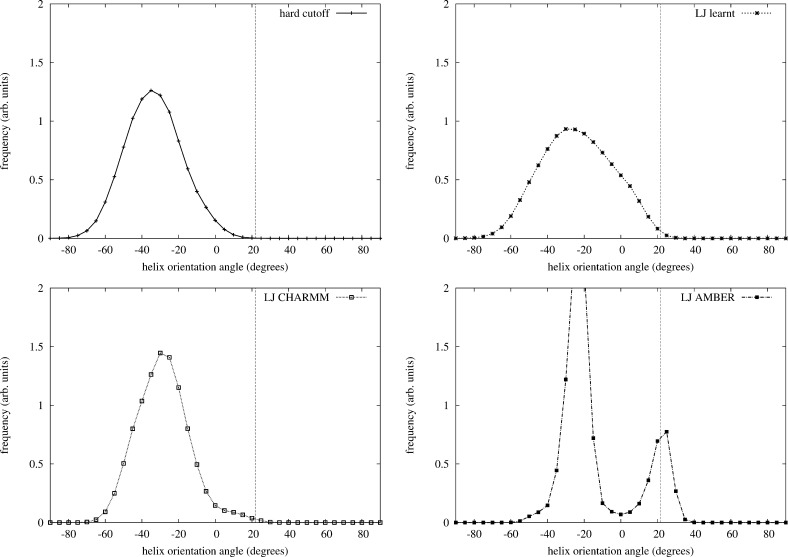
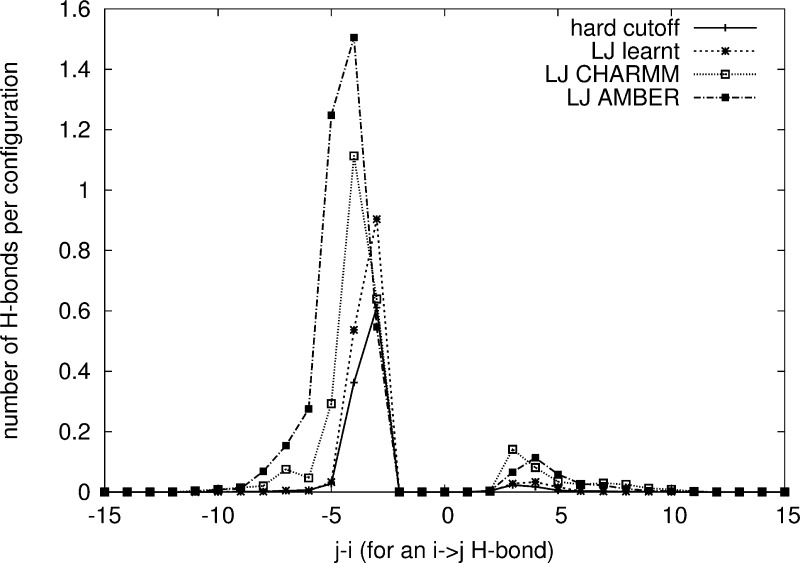
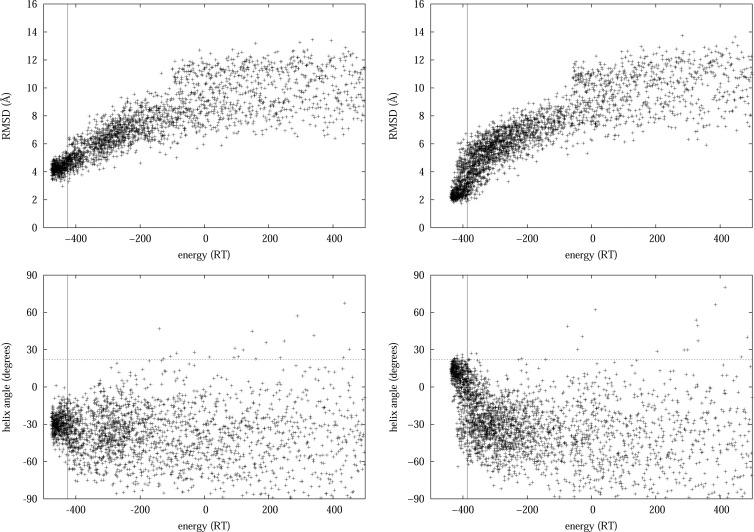
Maximum Likelihood (ML) optimization schemes are widely used for parameter inference. They maximize the likelihood of some experimentally observed data, with respect to the model parameters iteratively, following the gradient of the logarithm of the likelihood. Here, we employ a ML inference scheme to infer a generalizable, physics-based coarse-grained protein model (which includes Go̅-like biasing terms to stabilize secondary structure elements in room-temperature simulations), using native conformations of a training set of proteins as the observed data. Contrastive divergence, a novel statistical machine learning technique, is used to efficiently approximate the direction of the gradient ascent, which enables the use of a large training set of proteins. Unlike previous work, the generalizability of the protein model allows the folding of peptides and a protein (protein G) which are not part of the training set. We compare the same force field with different van der Waals (vdW) potential forms: a hard cutoff model, and a Lennard-Jones (LJ) potential with vdW parameters inferred or adopted from the CHARMM or AMBER force fields. Simulations of peptides and protein G show that the LJ model with inferred parameters outperforms the hard cutoff potential, which is consistent with previous observations. Simulations using the LJ potential with inferred vdW parameters also outperforms the protein models with adopted vdW parameter values, demonstrating that model parameters generally cannot be used with force fields with different energy functions. The software is available at https://sites.google.com/site/crankite/.
最大似然(ML)优化方案被广泛用于参数推断。它们通过迭代地遵循似然对数的梯度,使一些实验观测数据相对于模型参数的似然最大化。在这里,我们采用一种ML推断方案,以一组训练蛋白质的天然构象作为观测数据,来推断一个可推广的、基于物理的粗粒度蛋白质模型(该模型包括类似Go̅的偏置项,以在室温模拟中稳定二级结构元件)。对比散度,一种新颖的统计机器学习技术,被用于有效地近似梯度上升的方向,这使得能够使用大量的蛋白质训练集。与之前的工作不同,该蛋白质模型的可推广性使得不属于训练集的肽和一种蛋白质(蛋白质G)能够折叠。我们比较了具有不同范德华(vdW)势形式的相同力场:一个硬截止模型,以及一个具有从CHARMM或AMBER力场推断或采用的vdW参数的 Lennard-Jones(LJ)势。肽和蛋白质G的模拟表明,具有推断参数的LJ模型优于硬截止势,这与之前的观察结果一致。使用具有推断vdW参数的LJ势进行的模拟也优于采用vdW参数值的蛋白质模型,这表明模型参数通常不能与具有不同能量函数的力场一起使用。该软件可在https://sites.google.com/site/crankite/获取。