Kwok Lai-yu, Zhang Jiachao, Guo Zhuang, Gesudu Qimu, Zheng Yi, Qiao Jianmin, Huo Dongxue, Zhang Heping
Key Laboratory of Dairy Biotechnology and Bioengineering, Education Ministry of P. R. China, Department of Food Science and Engineering, Inner Mongolia Agricultural University, Hohhot, Inner Mongolia, P. R. China.
PLoS One. 2014 Apr 3;9(4):e93631. doi: 10.1371/journal.pone.0093631. eCollection 2014.
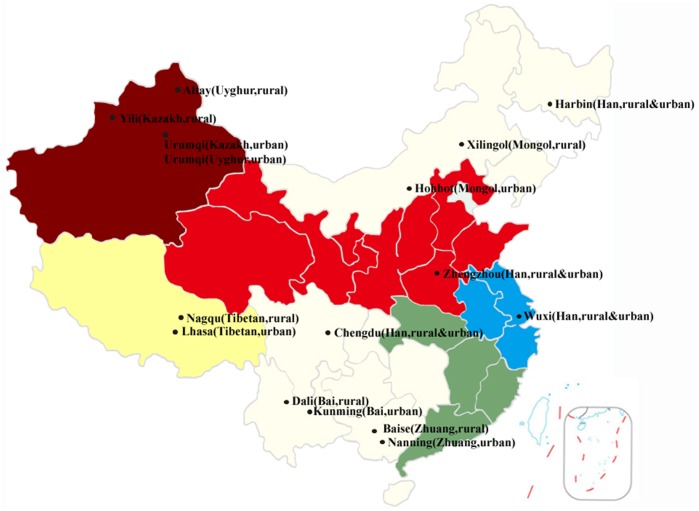
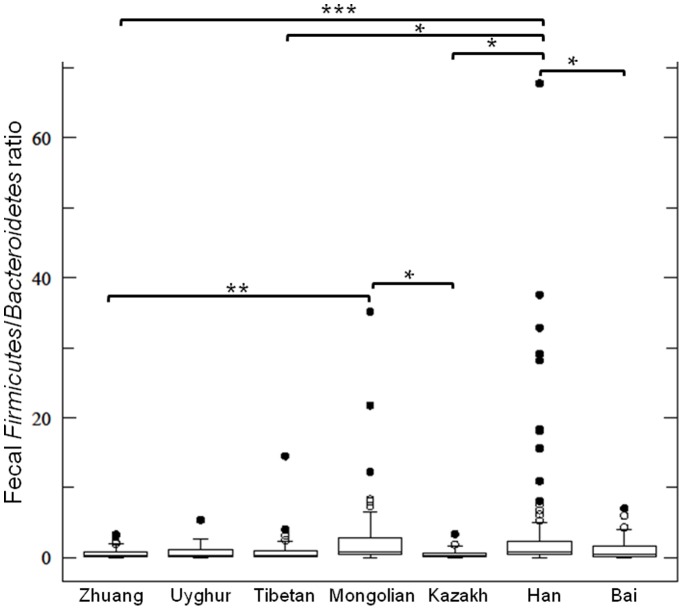
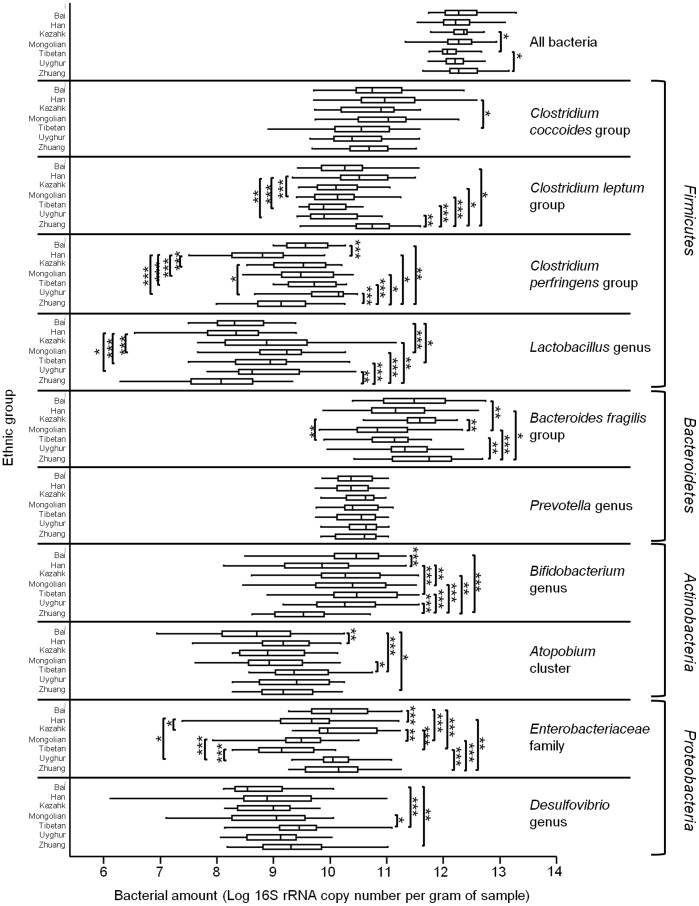
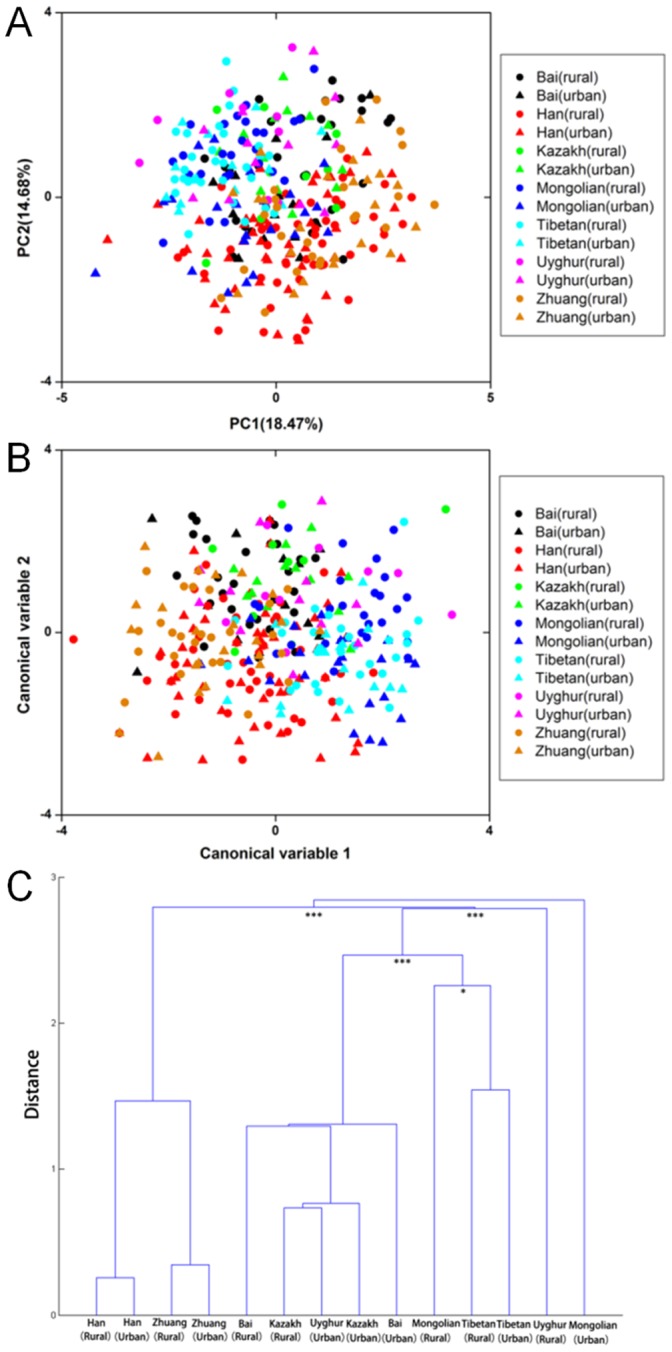
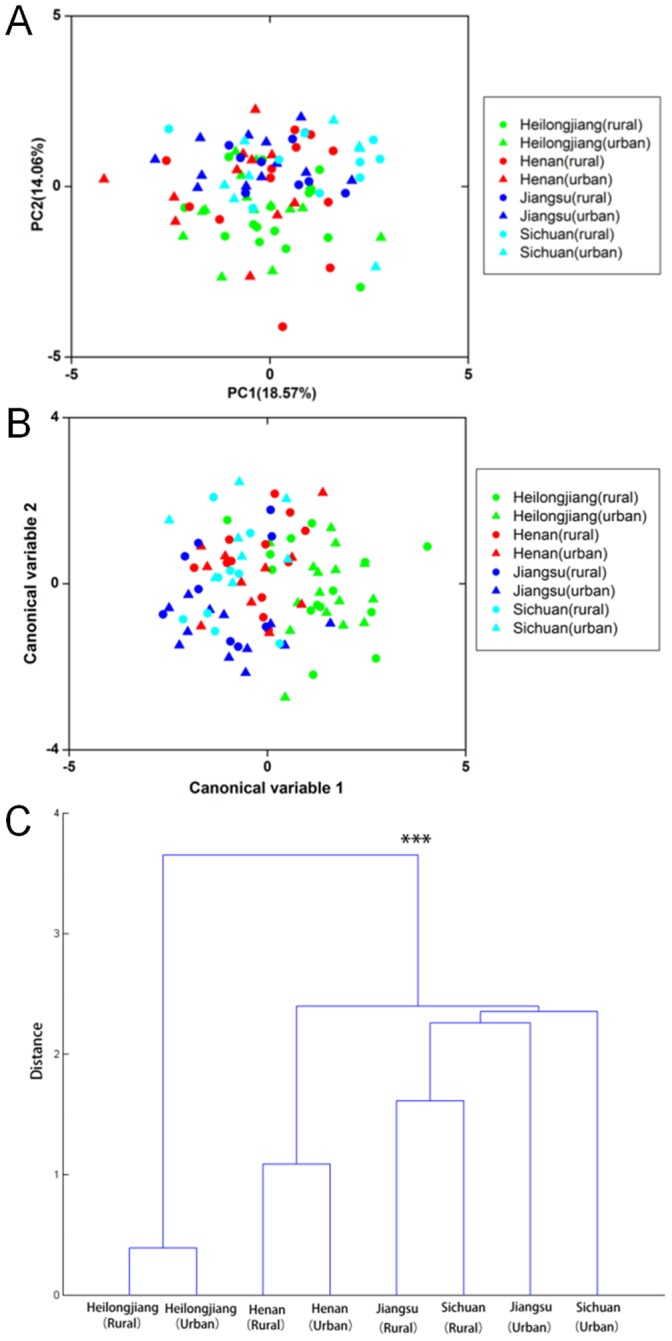
The human gut microbiota consists of complex microbial communities, which possibly play crucial roles in physiological functioning and health maintenance. China has evolved into a multicultural society consisting of the major ethnic group, Han, and 55 official ethnic minority groups. Nowadays, these minority groups inhabit in different Chinese provinces and some of them still keep their unique culture and lifestyle. Currently, only limited data are available on the gut microbiota of these Chinese ethnic groups. In this study, 10 major fecal bacterial groups of 314 healthy individuals from 7 Chinese ethnic origins were enumerated by quantitative polymerase chain reaction. Our data confirmed that the selected bacterial groups were common to all 7 surveyed ethnicities, but the amount of the individual bacterial groups varied to different degree. By principal component and canonical variate analyses of the 314 individuals or the 91 Han subjects, no distinct group clustering pattern was observed. Nevertheless, weak differences were noted between the Han and Zhuang from other ethnic minority groups, and between the Heilongjiang Hans from those of the other provinces. Thus, our results suggest that the ethnic origin may contribute to shaping the human gut microbiota.
人类肠道微生物群由复杂的微生物群落组成,这些群落可能在生理功能和健康维持中发挥关键作用。中国已发展成为一个多元文化社会,主要民族为汉族,还有55个法定少数民族。如今,这些少数民族居住在中国不同省份,其中一些仍保留着独特的文化和生活方式。目前,关于这些中国少数民族肠道微生物群的数据有限。在本研究中,通过定量聚合酶链反应对来自7个中国民族的314名健康个体的10种主要粪便细菌群进行了计数。我们的数据证实,所选细菌群在所有7个被调查民族中都很常见,但各个细菌群的数量在不同程度上有所差异。通过对314名个体或91名汉族受试者进行主成分分析和典型变量分析,未观察到明显的群体聚类模式。然而,汉族与其他少数民族中的壮族之间,以及黑龙江省汉族与其他省份汉族之间存在细微差异。因此,我们的结果表明,民族起源可能有助于塑造人类肠道微生物群。