Pressé Steve, Peterson Jack, Lee Julian, Elms Phillip, MacCallum Justin L, Marqusee Susan, Bustamante Carlos, Dill Ken
Department of Physics, Indiana University-Purdue University , Indianapolis, Indiana 46202, United States.
J Phys Chem B. 2014 Jun 19;118(24):6597-603. doi: 10.1021/jp500611f. Epub 2014 Jun 5.
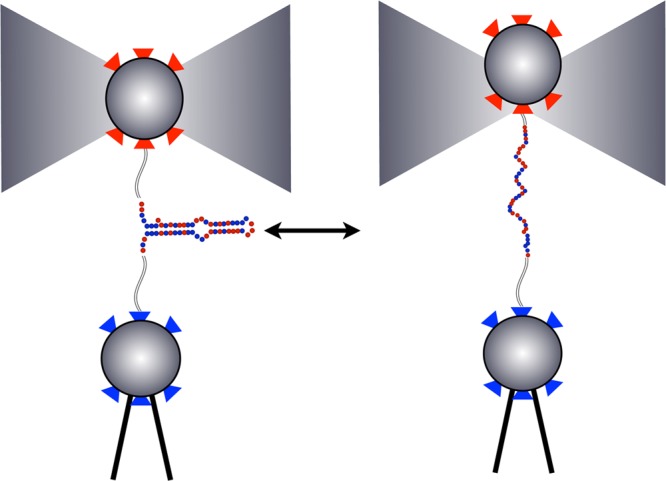
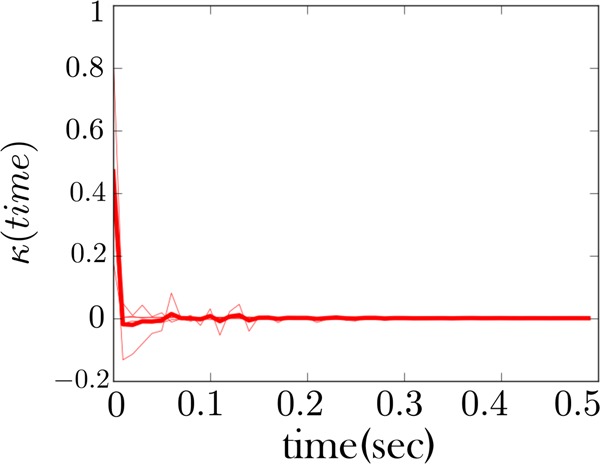
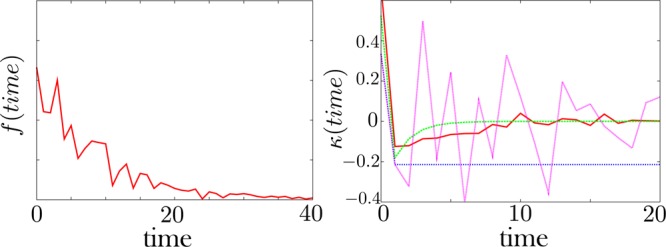
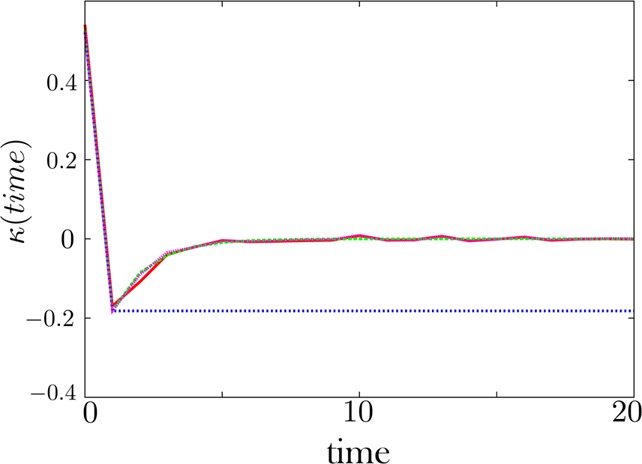
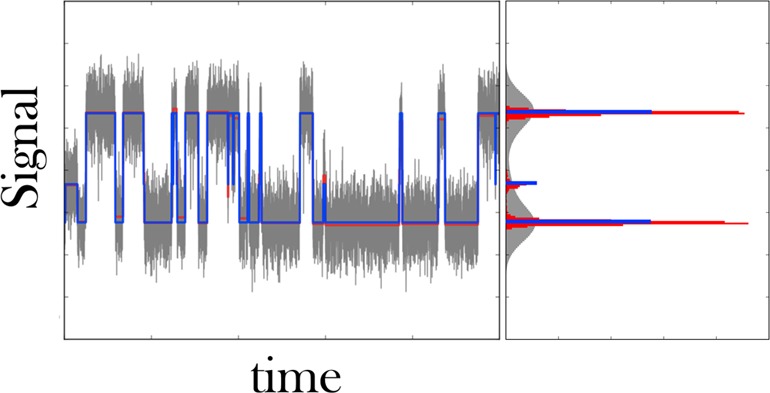
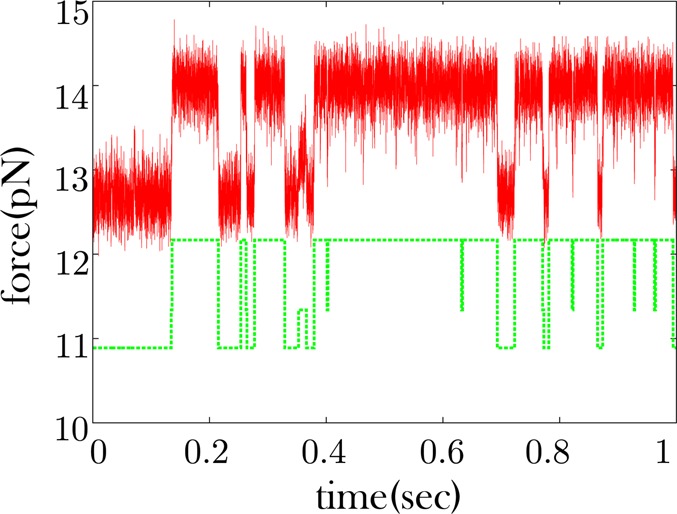
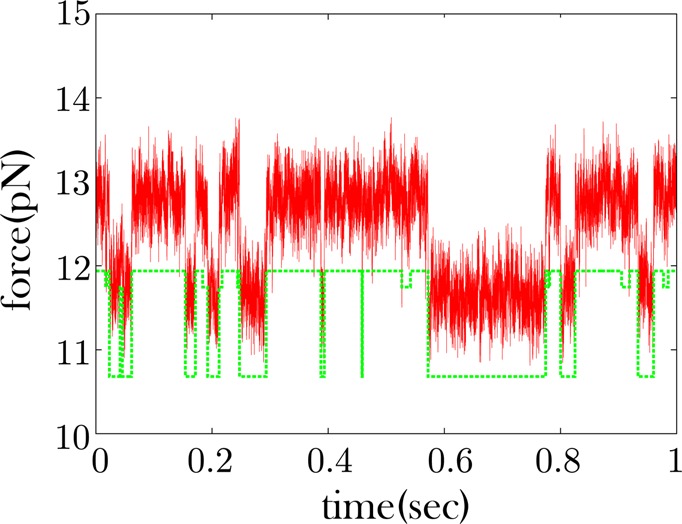
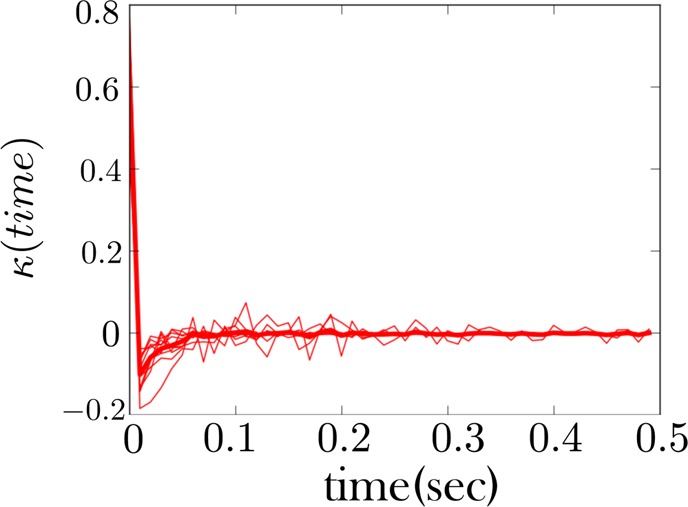
Extracting kinetic models from single molecule data is an important route to mechanistic insight in biophysics, chemistry, and biology. Data collected from force spectroscopy can probe discrete hops of a single molecule between different conformational states. Model extraction from such data is a challenging inverse problem because single molecule data are noisy and rich in structure. Standard modeling methods normally assume (i) a prespecified number of discrete states and (ii) that transitions between states are Markovian. The data set is then fit to this predetermined model to find a handful of rates describing the transitions between states. We show that it is unnecessary to assume either (i) or (ii) and focus our analysis on the zipping/unzipping transitions of an RNA hairpin. The key is in starting with a very broad class of non-Markov models in order to let the data guide us toward the best model from this very broad class. Our method suggests that there exists a folding intermediate for the P5ab RNA hairpin whose zipping/unzipping is monitored by force spectroscopy experiments. This intermediate would not have been resolved if a Markov model had been assumed from the onset. We compare the merits of our method with those of others.
从单分子数据中提取动力学模型是深入了解生物物理学、化学和生物学机制的重要途径。通过力谱法收集的数据可以探测单个分子在不同构象状态之间的离散跳跃。从这类数据中提取模型是一个具有挑战性的反问题,因为单分子数据存在噪声且结构丰富。标准建模方法通常假设:(i)离散状态的数量预先确定;(ii)状态之间的转变是马尔可夫的。然后将数据集拟合到这个预先确定的模型,以找到少数几个描述状态之间转变的速率。我们表明,既不需要假设(i)也不需要假设(ii),并将我们的分析重点放在RNA发夹的拉链化/解拉链化转变上。关键在于从一类非常广泛的非马尔可夫模型入手,以便让数据引导我们从这个非常广泛的类别中找到最佳模型。我们的方法表明,P5ab RNA发夹存在一个折叠中间体,其拉链化/解拉链化过程由力谱实验监测。如果从一开始就假设是马尔可夫模型,这个中间体就不会被解析出来。我们将我们的方法与其他方法的优点进行了比较。