Bian Zhiguo, Marvin Christopher C, Pettersson Martin, Martin Stephen F
Department of Chemistry, The University of Texas , Austin, Texas 78712, United States.
J Am Chem Soc. 2014 Oct 8;136(40):14184-92. doi: 10.1021/ja5074646. Epub 2014 Sep 29.
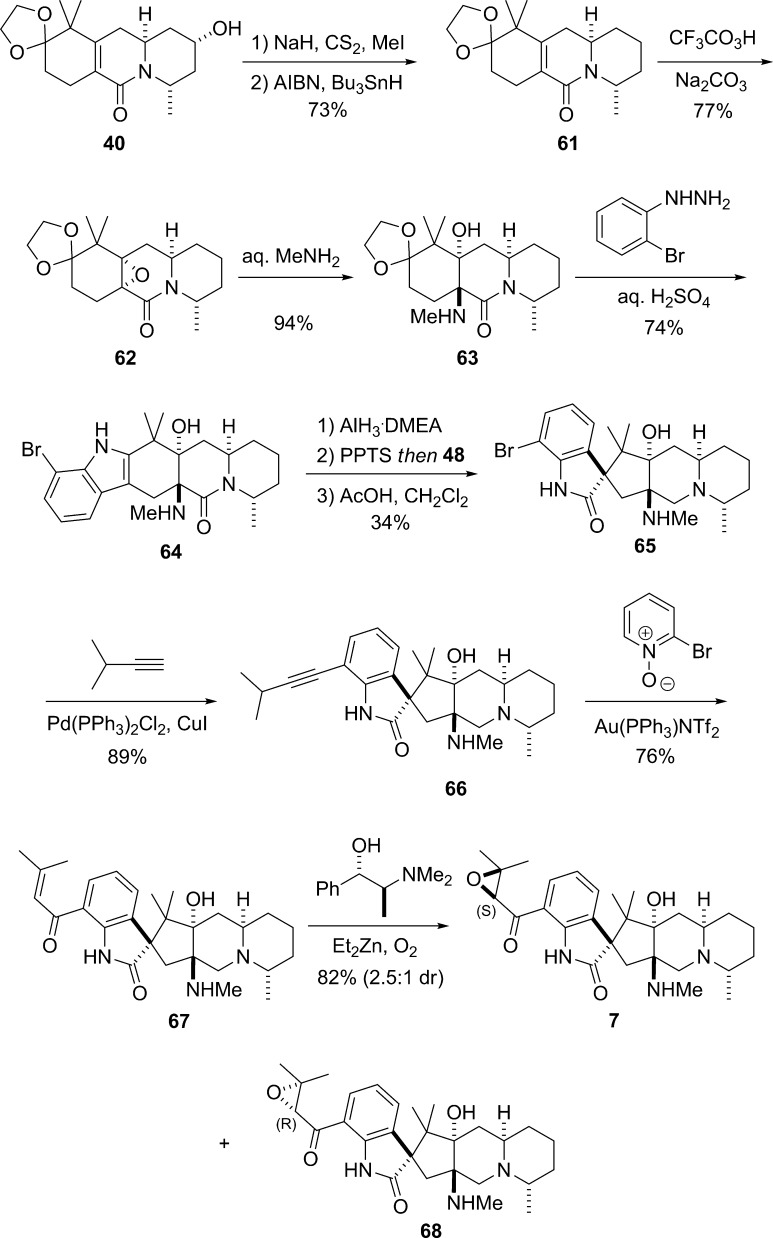
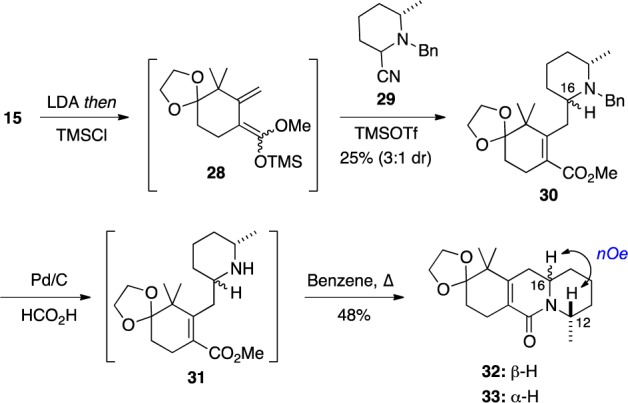
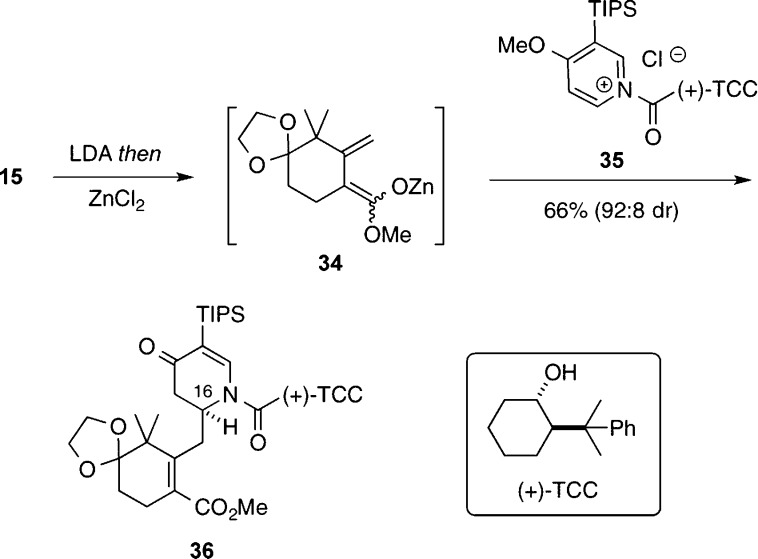
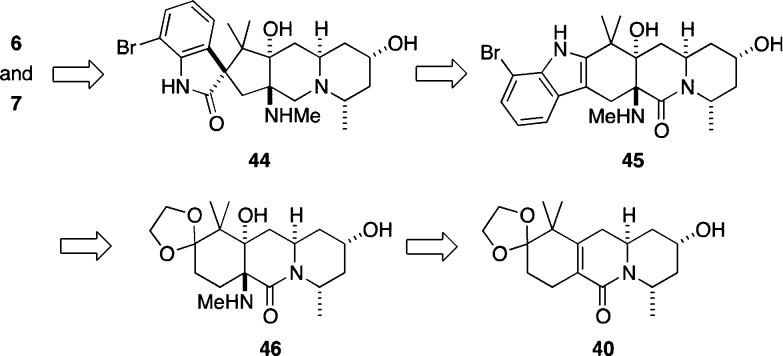
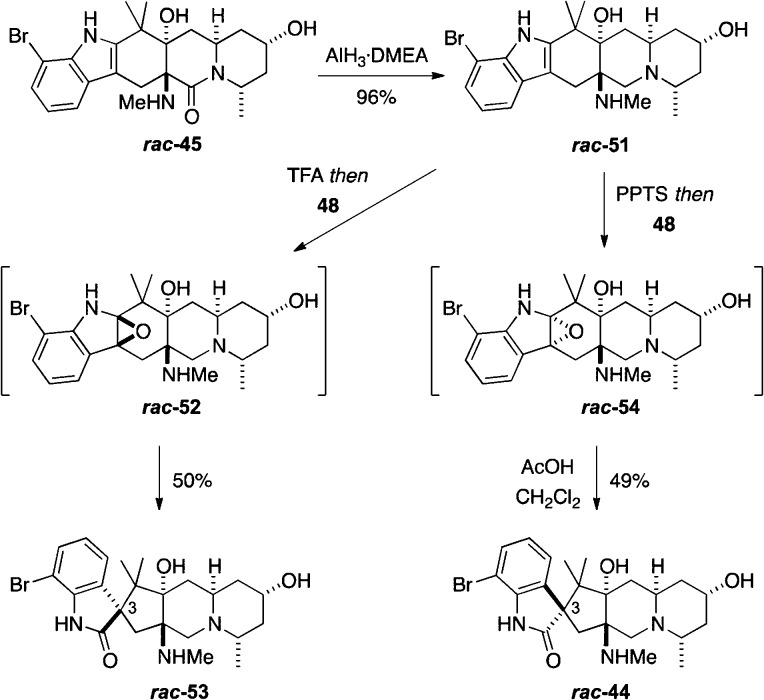
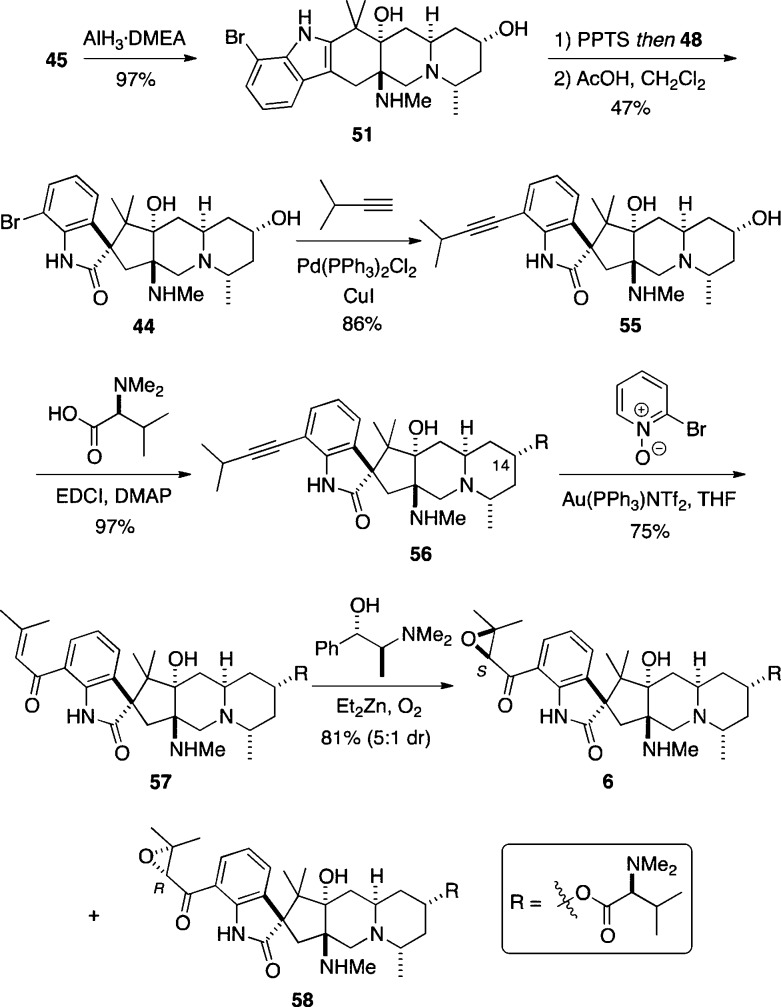
The concise, enantioselective total syntheses of (-)-citrinadin A and (+)-citrinadin B in a total of only 20 and 21 steps, respectively, from commercially available starting materials are described. Our strategy, which minimizes refunctionalization and protection/deprotection operations, features the highly diastereoselective, vinylogous Mannich addition of a dienolate to a chiral pyridinium salt to set the first chiral center. The absolute stereochemistry of this key center was then relayed by a sequence of substrate-controlled reactions, including a highly stereoselective epoxidation/ring opening sequence and an oxidative rearrangement of an indole to furnish a spirooxindole to establish the remaining stereocenters in the pentacyclic core of the citrinadins. An early stage intermediate in the synthesis of (-)-citrinadin A was deoxygenated to generate a dehydroxy compound that was elaborated into (+)-citrinadin B by a sequence of reaction identical to those used to prepare (-)-citrinadin A. These concise syntheses of (-)-citrinadin A and (+)-citrinadin B led to a revision of their stereochemical structures.
本文描述了分别以市售起始原料,仅通过20步和21步的简洁对映选择性全合成方法,合成(-)-柑橘霉素A和(+)-柑橘霉素B。我们的策略是尽量减少重新官能团化和保护/脱保护操作,其特点是将烯醇化物高度非对映选择性地、乙烯型曼尼希加成到手性吡啶盐上,以构建第一个手性中心。然后通过一系列底物控制的反应传递该关键中心的绝对立体化学,包括高度立体选择性的环氧化/开环序列以及吲哚的氧化重排,以提供一个螺环氧化吲哚,从而在柑橘霉素的五环核心中建立其余的立体中心。在合成(-)-柑橘霉素A的早期中间体被脱氧生成一种脱羟基化合物,该化合物通过与制备(-)-柑橘霉素A相同的一系列反应转化为(+)-柑橘霉素B。(-)-柑橘霉素A和(+)-柑橘霉素B的这些简洁合成导致了它们立体化学结构的修正。