Park HeeKuk, Shin Jong Wook, Park Sang-Gue, Kim Wonyong
Department of Microbiology, College of Medicine, Chung-Ang University, Seoul, South Korea.
Division of Pulmonology and Allergology, Department of Internal Medicine, College of Medicine, Chung-Ang University, Seoul, South Korea.
PLoS One. 2014 Oct 16;9(10):e109710. doi: 10.1371/journal.pone.0109710. eCollection 2014.
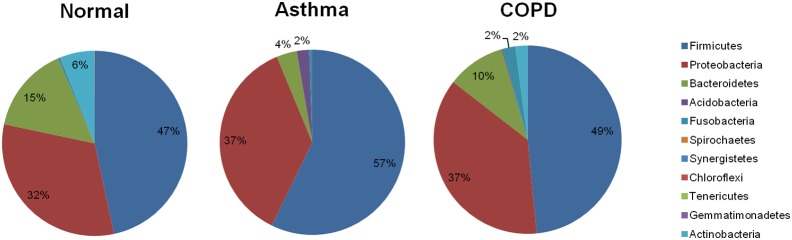
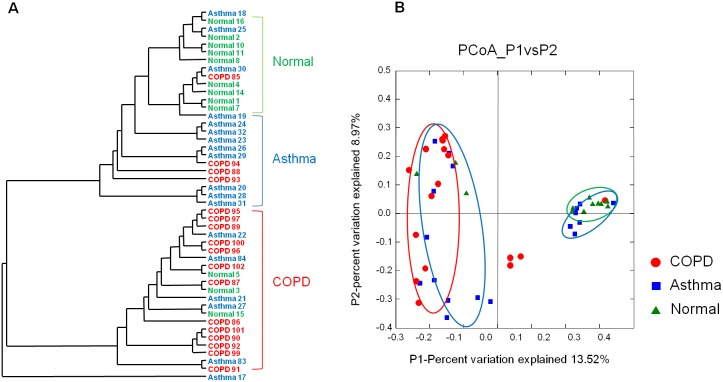
Respiratory infections are well-known triggers of chronic respiratory diseases. Recently, culture-independent tools have indicated that lower airway microbiota may contribute to pathophysiologic processes associated with asthma and chronic obstructive pulmonary disease (COPD). However, the relationship between upper airway microbiota and chronic respiratory diseases remains unclear. This study was undertaken to define differences of microbiota in the oropharynx of asthma and COPD patients relative to those in healthy individuals. To account for the qualitative and quantitative diversity of the 16S rRNA gene in the oropharynx, the microbiomes of 18 asthma patients, 17 COPD patients, and 12 normal individuals were assessed using a high-throughput next-generation sequencing analysis. In the 259,572 total sequence reads, α and β diversity measurements and a generalized linear model revealed that the oropharynx microbiota are diverse, but no significant differences were observed between asthma and COPD patients. Pseudomonas spp. of Proteobacteria and Lactobacillus spp. of Firmicutes were highly abundant in asthma and COPD. By contrast, Streptococcus, Veillonella, Prevotella, and Neisseria of Bacteroidetes dominated in the healthy oropharynx. These findings are consistent with previous studies conducted in the lower airways and suggest that oropharyngeal airway microbiota are important for understanding the relationships between the various parts of the respiratory tract with regard to bacterial colonization and comprehensive assessment of asthma and COPD.
呼吸道感染是慢性呼吸道疾病众所周知的诱因。最近,不依赖培养的工具表明,下呼吸道微生物群可能参与了与哮喘和慢性阻塞性肺疾病(COPD)相关的病理生理过程。然而,上呼吸道微生物群与慢性呼吸道疾病之间的关系仍不清楚。本研究旨在确定哮喘和COPD患者口咽部微生物群与健康个体口咽部微生物群的差异。为了说明口咽部16S rRNA基因的定性和定量多样性,使用高通量下一代测序分析评估了18例哮喘患者、17例COPD患者和12例正常个体的微生物组。在总共259,572条序列读数中,α和β多样性测量以及广义线性模型显示口咽部微生物群具有多样性,但哮喘患者和COPD患者之间未观察到显著差异。变形菌门的假单胞菌属和厚壁菌门的乳杆菌属在哮喘和COPD中含量很高。相比之下,拟杆菌门的链球菌属、韦荣球菌属、普雷沃菌属和奈瑟菌属在健康口咽部占主导地位。这些发现与之前在下呼吸道进行的研究一致,表明口咽气道微生物群对于理解呼吸道各部分之间关于细菌定植的关系以及哮喘和COPD的综合评估很重要。