Shteynberg David, Mendoza Luis, Hoopmann Michael R, Sun Zhi, Schmidt Frank, Deutsch Eric W, Moritz Robert L
Institute for Systems Biology, Seattle, WA, USA.
ZIK-FunGene Junior Research Group Applied Proteomics, Interfaculty Institute for Genetics and Functional Genomics, University Medicine Greifswald, Greifswald, Germany.
J Am Soc Mass Spectrom. 2015 Nov;26(11):1837-47. doi: 10.1007/s13361-015-1252-5. Epub 2015 Sep 29.
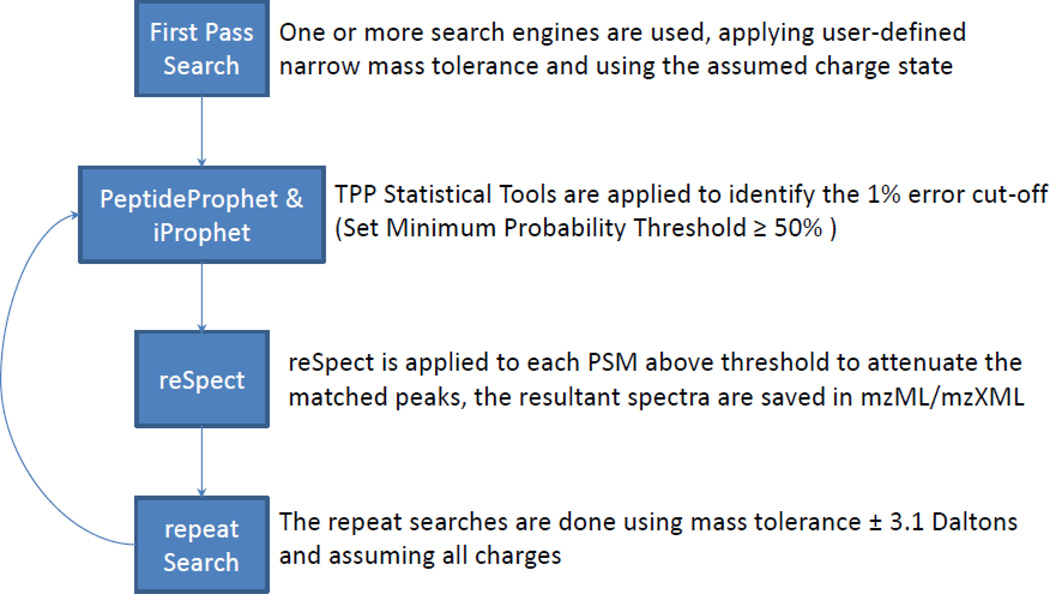
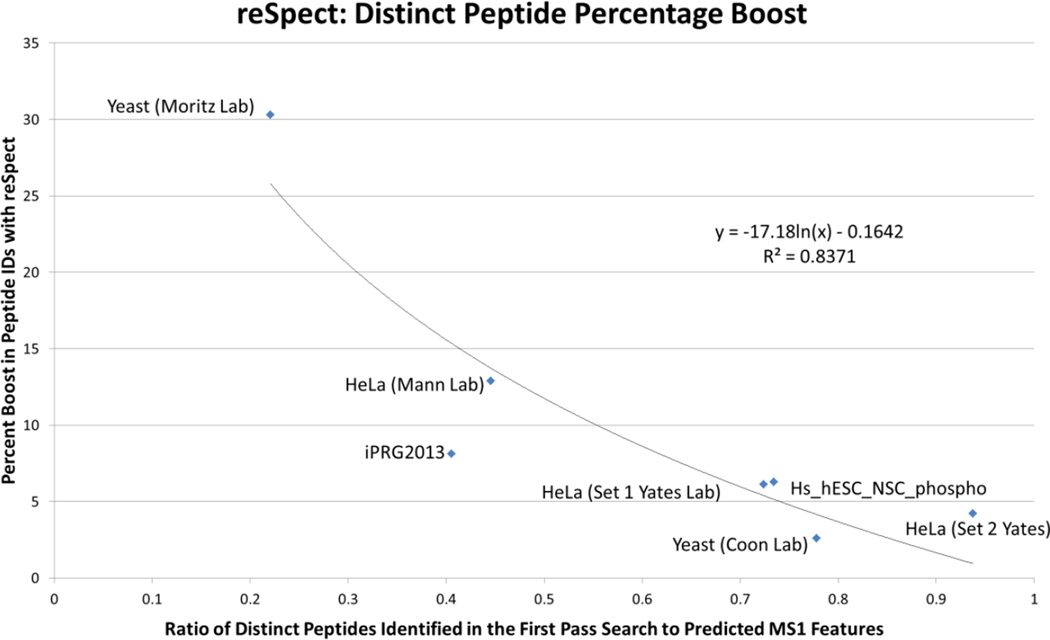
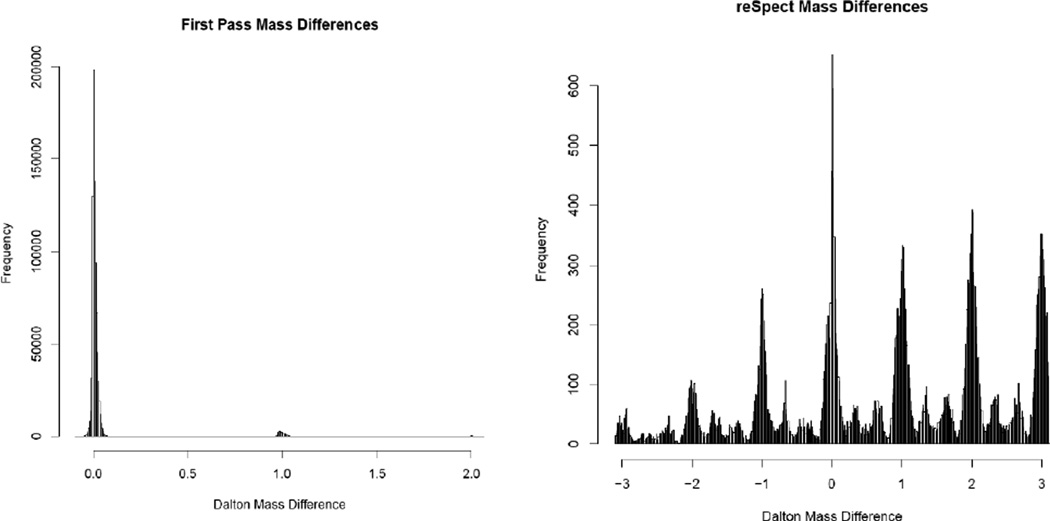
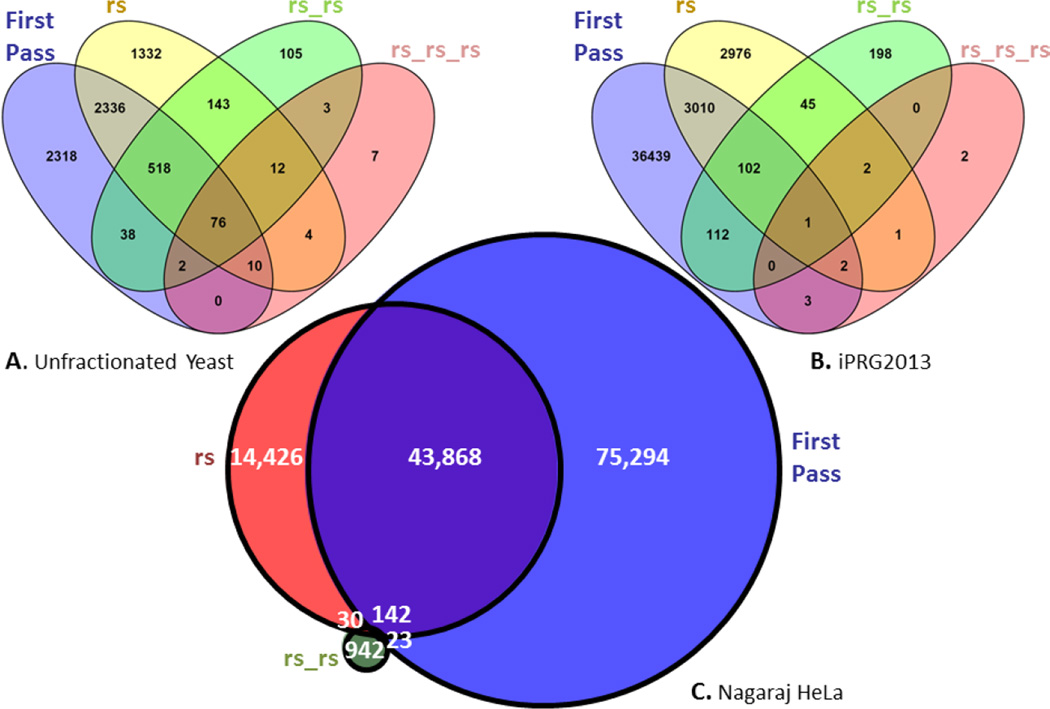
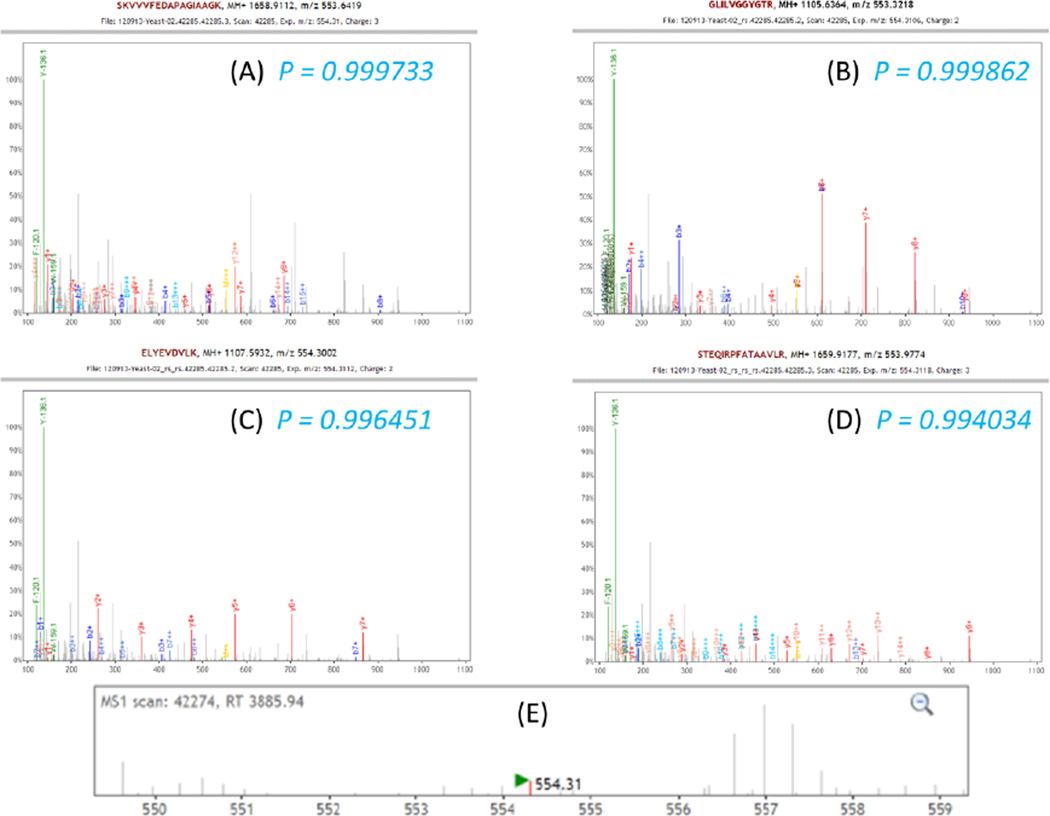
Most shotgun proteomics data analysis workflows are based on the assumption that each fragment ion spectrum is explained by a single species of peptide ion isolated by the mass spectrometer; however, in reality mass spectrometers often isolate more than one peptide ion within the window of isolation that contribute to additional peptide fragment peaks in many spectra. We present a new tool called reSpect, implemented in the Trans-Proteomic Pipeline (TPP), which enables an iterative workflow whereby fragment ion peaks explained by a peptide ion identified in one round of sequence searching or spectral library search are attenuated based on the confidence of the identification, and then the altered spectrum is subjected to further rounds of searching. The reSpect tool is not implemented as a search engine, but rather as a post-search engine processing step where only fragment ion intensities are altered. This enables the application of any search engine combination in the iterations that follow. Thus, reSpect is compatible with all other protein sequence database search engines as well as peptide spectral library search engines that are supported by the TPP. We show that while some datasets are highly amenable to chimeric spectrum identification and lead to additional peptide identification boosts of over 30% with as many as four different peptide ions identified per spectrum, datasets with narrow precursor ion selection only benefit from such processing at the level of a few percent. We demonstrate a technique that facilitates the determination of the degree to which a dataset would benefit from chimeric spectrum analysis. The reSpect tool is free and open source, provided within the TPP and available at the TPP website. Graphical Abstract ᅟ.
每个碎片离子谱由质谱仪分离出的单一肽离子种类来解释;然而,实际上质谱仪在隔离窗口内常常分离出不止一种肽离子,这在许多谱图中导致了额外的肽片段峰。我们展示了一种名为reSpect的新工具,它在跨蛋白质组学管道(TPP)中实现,支持一种迭代工作流程,即根据一轮序列搜索或谱图库搜索中鉴定出的肽离子所解释的碎片离子峰,基于鉴定的置信度进行衰减,然后将改变后的谱图进行进一步的搜索轮次。reSpect工具并非作为搜索引擎实现,而是作为搜索后引擎处理步骤,仅改变碎片离子强度。这使得在后续迭代中能够应用任何搜索引擎组合。因此,reSpect与TPP支持的所有其他蛋白质序列数据库搜索引擎以及肽谱图库搜索引擎兼容。我们表明,虽然一些数据集非常适合嵌合谱图鉴定,并且每个谱图能鉴定多达四个不同肽离子时可使额外肽鉴定增加超过30%,但仅具有窄前体离子选择的数据集仅能从这种处理中获得几个百分点的益处。我们展示了一种有助于确定数据集从嵌合谱图分析中受益程度的技术。reSpect工具是免费且开源的,包含在TPP中,可在TPP网站获取。图形摘要ᅟ。