Momeni Stephanie S, Whiddon Jennifer, Cheon Kyounga, Moser Stephen A, Childers Noel K
Department of Pediatric Dentistry, School of Dentistry, University of Alabama at Birmingham, SOD 304, 1720 2nd Avenue South, Birmingham, AL 35294-0007, USA.
Department of Pathology, School of Medicine & Dentistry, University of Alabama at Birmingham, WP 230, 619 19th Street South, Birmingham, AL 35249-7331, USA.
Arch Oral Biol. 2015 Dec;60(12):1769-76. doi: 10.1016/j.archoralbio.2015.09.012. Epub 2015 Sep 21.
Two multilocus sequencing typing (MLST) schemes are currently available for Streptococcus mutans. The first, introduced by Nakano et al. in 2007, consists of 8 conserved housekeeping genes. The second, introduced in 2010 by Do et al., includes 6 housekeeping genes and 2 putative virulence genes. The purpose of the current study was to compare the two MLST schemes for use in validating repetitive extragenic palindromic polymerase chain reaction (rep-PCR) genotypes.
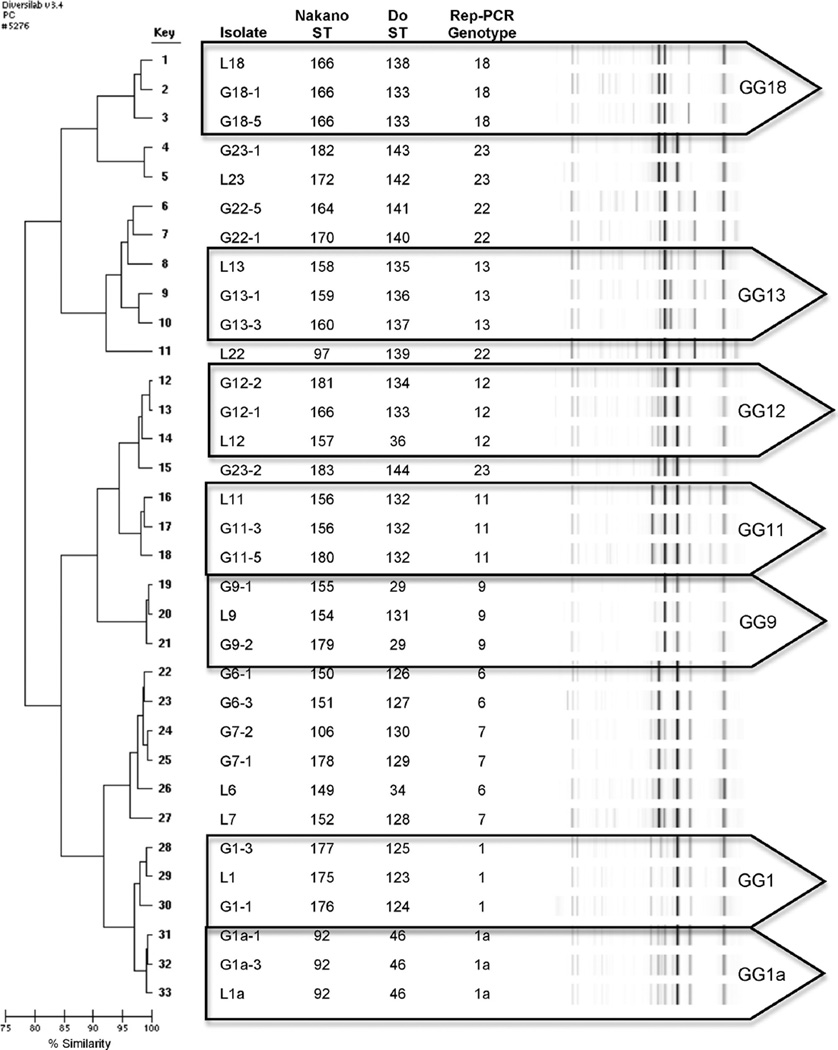
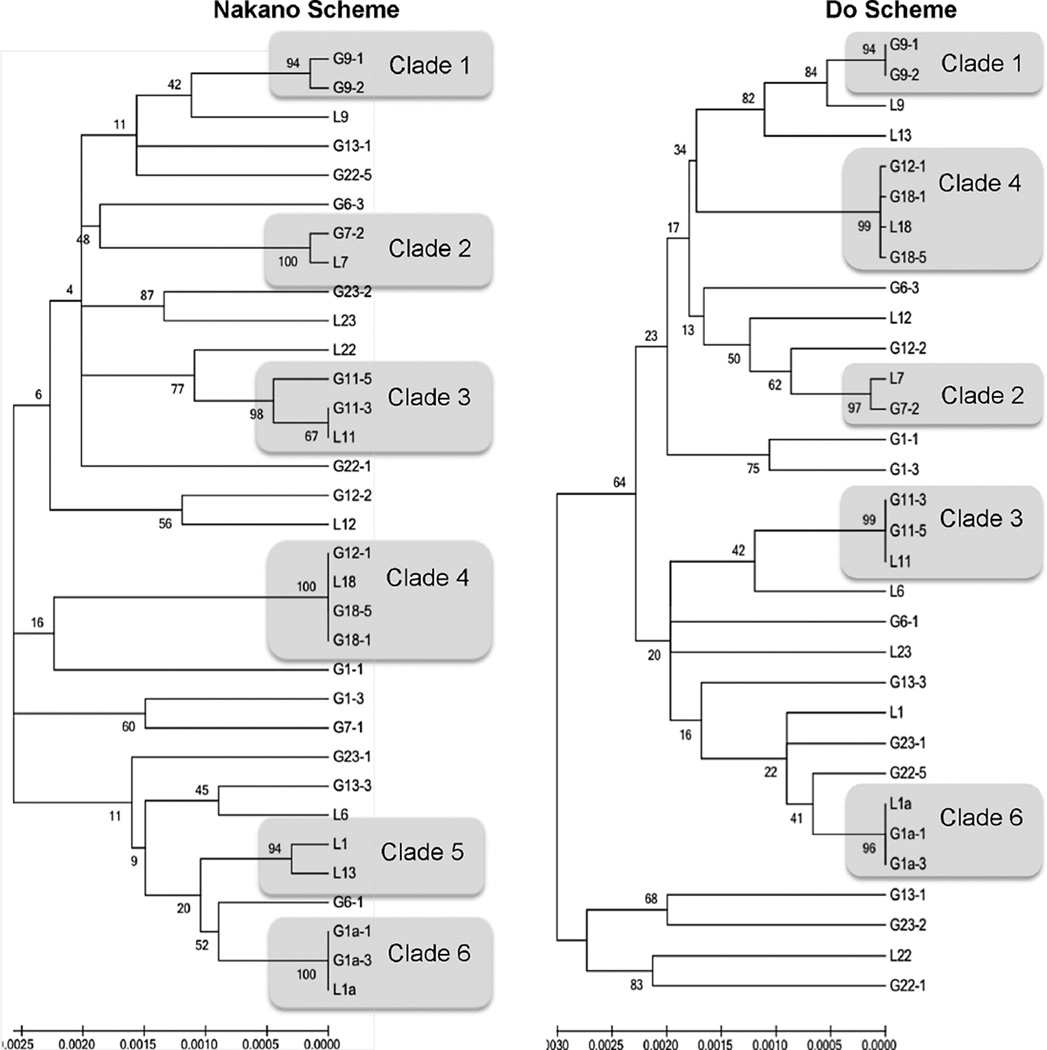
Thirty-three S. mutans isolates, representing the 11 most commonly occurring rep-PCR genotype groups, were selected for MLST. MLST was performed with SYBR Green™ PCR with published primers for both MLST schemes. Amplicons were purified, sequenced, and data checked against the www.PubMLST.org database for allelic and sequence type (ST) assignment. Discriminatory power, congruence, and convenience criteria were evaluated. Concatenated sequences for each scheme were analyzed using MEGA to generate phylogenetic trees using minimum evolution with bootstrap.
No significant difference in discriminatory power was observed between the two MLST schemes for S. mutans. Clonal clusters were consistent for both schemes. Overall, MLST demonstrated marginally greater discriminatory power than rep-PCR; however all methods were found to be congruent. New alleles and ST are reported for each scheme and added to the PubMLST database.
Clonality, supported by both methods and rep-PCR, indicates S. mutans genotypes are shared between unrelated subjects. Both Nakano and Do schemes demonstrates similar genotype discrimination for S. mutans isolates suggesting each are well designed and may be used to verify rep-PCR genotypes.
目前有两种多位点测序分型(MLST)方案可用于变形链球菌。第一种由中野等人于2007年提出,由8个保守的管家基因组成。第二种由多等人于2010年提出,包括6个管家基因和2个假定的毒力基因。本研究的目的是比较这两种MLST方案在验证重复外显子回文聚合酶链反应(rep-PCR)基因型中的应用。
选择代表11个最常见的rep-PCR基因型组的33株变形链球菌分离株进行MLST。两种MLST方案均使用SYBR Green™PCR和已发表的引物进行MLST。扩增子经纯化、测序,并根据www.PubMLST.org数据库检查数据以进行等位基因和序列类型(ST)分配。评估了鉴别力、一致性和便利性标准。使用MEGA分析每种方案的串联序列,以使用最小进化法和自展法生成系统发育树。
两种MLST方案对变形链球菌的鉴别力无显著差异。两种方案的克隆簇一致。总体而言,MLST的鉴别力略高于rep-PCR;然而,所有方法都被发现是一致的。报告了每种方案的新等位基因和ST,并添加到PubMLST数据库中。
两种方法和rep-PCR均支持的克隆性表明,变形链球菌基因型在不相关的个体之间共享。中野和多的方案对变形链球菌分离株的基因型鉴别相似,表明每种方案设计良好,可用于验证rep-PCR基因型。