Walzthoeni Thomas, Joachimiak Lukasz A, Rosenberger George, Röst Hannes L, Malmström Lars, Leitner Alexander, Frydman Judith, Aebersold Ruedi
Department of Biology, Eidgenössische Technische Hochschule (ETH) Zurich, Zurich, Switzerland.
Gene Center Munich, Ludwig-Maximilians-Universität München, Munich, Germany.
Nat Methods. 2015 Dec;12(12):1185-90. doi: 10.1038/nmeth.3631. Epub 2015 Oct 26.
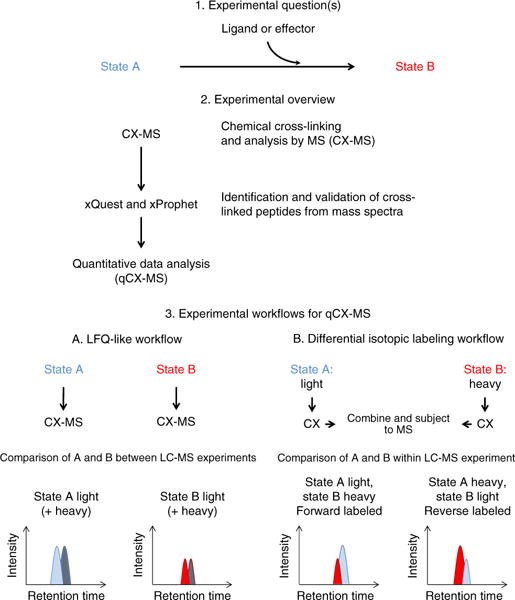
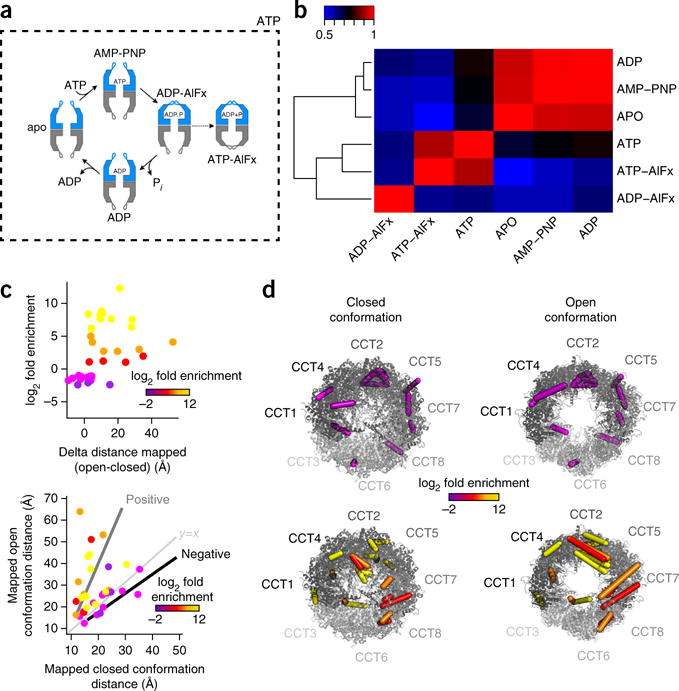
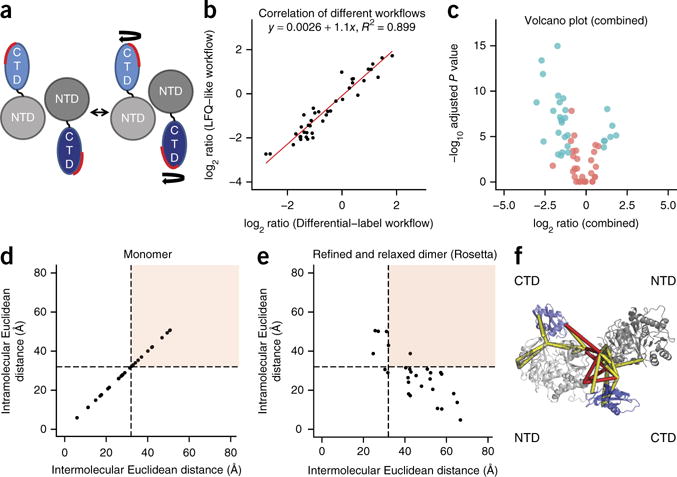
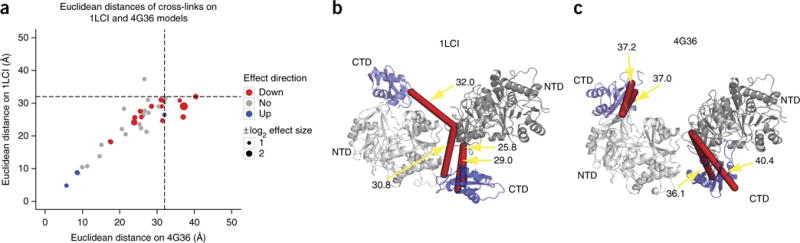
Chemical cross-linking in combination with mass spectrometry generates distance restraints of amino acid pairs in close proximity on the surface of native proteins and protein complexes. In this study we used quantitative mass spectrometry and chemical cross-linking to quantify differences in cross-linked peptides obtained from complexes in spatially discrete states. We describe a generic computational pipeline for quantitative cross-linking mass spectrometry consisting of modules for quantitative data extraction and statistical assessment of the obtained results. We used the method to detect conformational changes in two model systems: firefly luciferase and the bovine TRiC complex. Our method discovers and explains the structural heterogeneity of protein complexes using only sparse structural information.
化学交联与质谱联用可生成天然蛋白质和蛋白质复合物表面紧密相邻氨基酸对的距离限制。在本研究中,我们使用定量质谱和化学交联来量化从处于空间离散状态的复合物中获得的交联肽的差异。我们描述了一种用于定量交联质谱的通用计算流程,该流程由用于定量数据提取和对所得结果进行统计评估的模块组成。我们使用该方法检测了两个模型系统中的构象变化:萤火虫荧光素酶和牛TRiC复合物。我们的方法仅使用稀疏的结构信息就能发现并解释蛋白质复合物的结构异质性。