Song Fan, Li Hu, Jiang Pei, Zhou Xuguo, Liu Jinpeng, Sun Changhai, Vogler Alfried P, Cai Wanzhi
Department of Entomology, China Agricultural University, Beijing, China.
Department of Entomology, University of Kentucky, Lexington.
Genome Biol Evol. 2016 May 22;8(5):1411-26. doi: 10.1093/gbe/evw086.
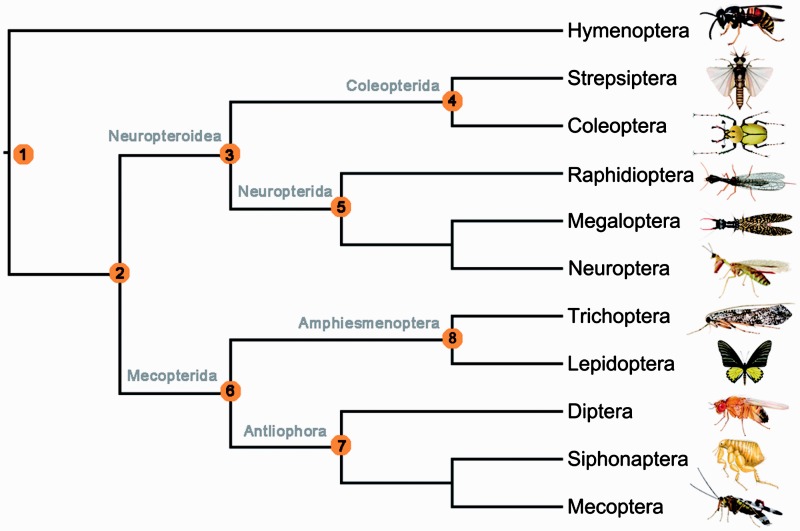
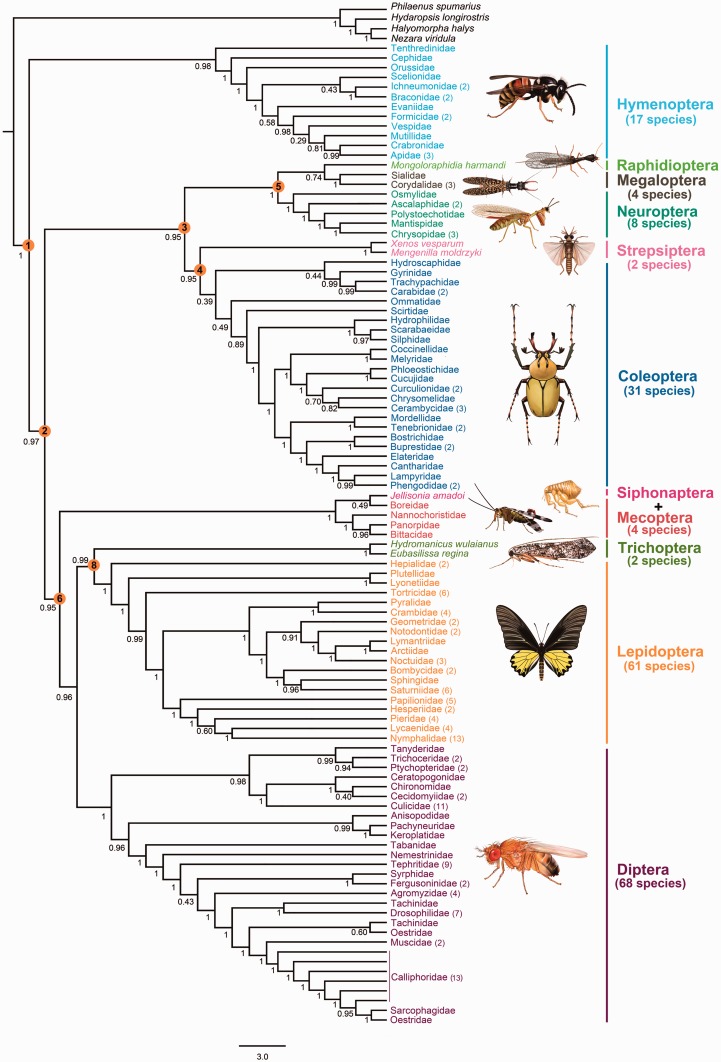
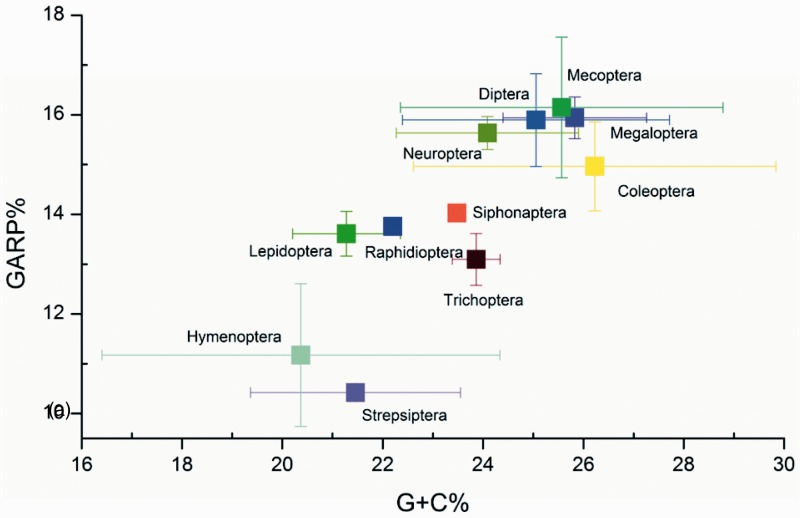
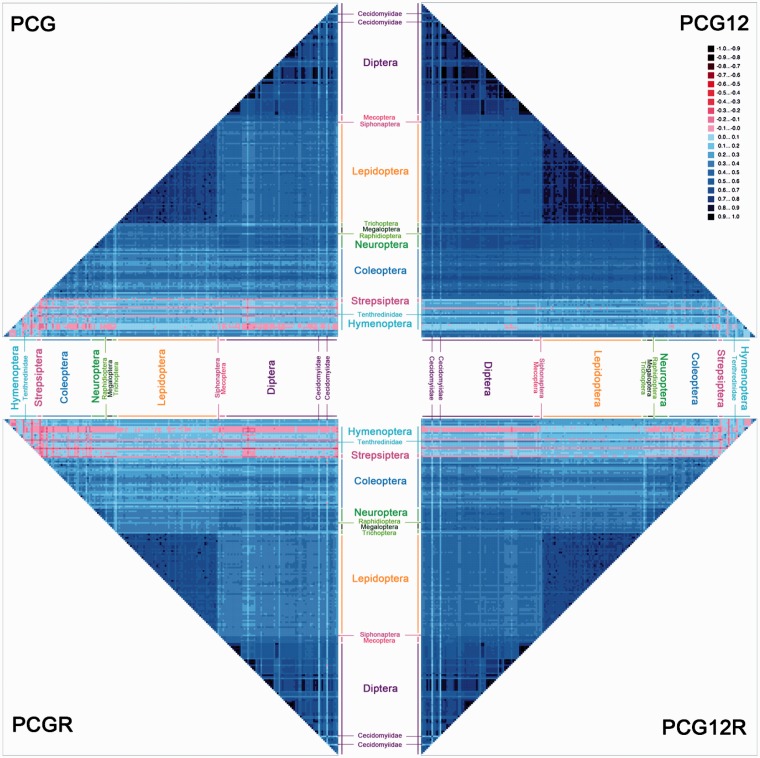
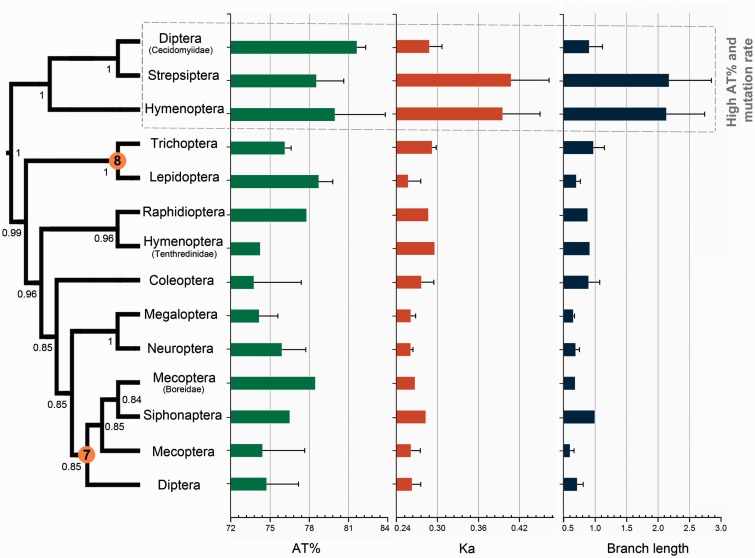
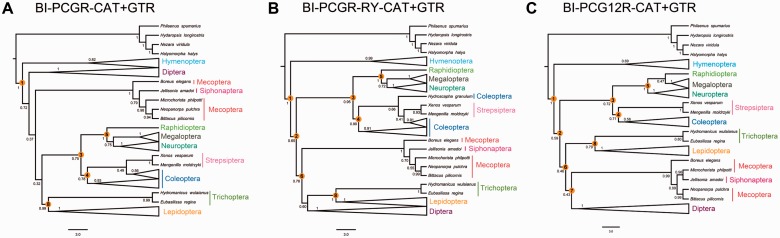
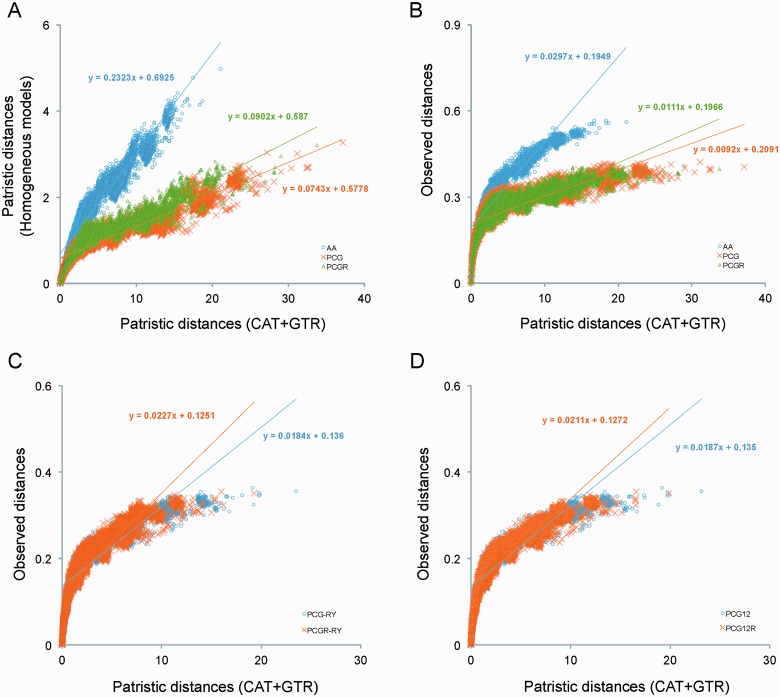
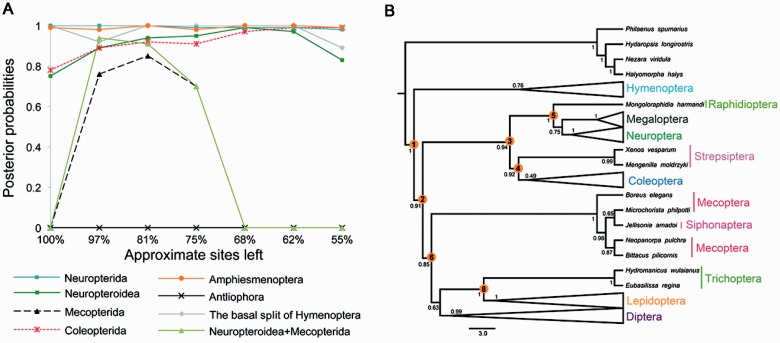
After decades of debate, a mostly satisfactory resolution of relationships among the 11 recognized holometabolan orders of insects has been reached based on nuclear genes, resolving one of the most substantial branches of the tree-of-life, but the relationships are still not well established with mitochondrial genome data. The main reasons have been the absence of sufficient data in several orders and lack of appropriate phylogenetic methods that avoid the systematic errors from compositional and mutational biases in insect mitochondrial genomes. In this study, we assembled the richest taxon sampling of Holometabola to date (199 species in 11 orders), and analyzed both nucleotide and amino acid data sets using several methods. We find the standard Bayesian inference and maximum-likelihood analyses were strongly affected by systematic biases, but the site-heterogeneous mixture model implemented in PhyloBayes avoided the false grouping of unrelated taxa exhibiting similar base composition and accelerated evolutionary rate. The inclusion of rRNA genes and removal of fast-evolving sites with the observed variability sorting method for identifying sites deviating from the mean rates improved the phylogenetic inferences under a site-heterogeneous model, correctly recovering most deep branches of the Holometabola phylogeny. We suggest that the use of mitochondrial genome data for resolving deep phylogenetic relationships requires an assessment of the potential impact of substitutional saturation and compositional biases through data deletion strategies and by using site-heterogeneous mixture models. Our study suggests a practical approach for how to use densely sampled mitochondrial genome data in phylogenetic analyses.
经过数十年的争论,基于核基因,已就昆虫11个公认的全变态目之间的关系达成了一个大体令人满意的解决方案,解决了生命之树中一个最重要的分支问题,但线粒体基因组数据仍未很好地确立这些关系。主要原因是几个目的数据不足,以及缺乏适当的系统发育方法来避免昆虫线粒体基因组中组成和突变偏差导致的系统误差。在本研究中,我们汇集了迄今为止最丰富的全变态类分类群样本(11个目中的199个物种),并使用多种方法分析了核苷酸和氨基酸数据集。我们发现标准的贝叶斯推断和最大似然分析受到系统偏差的强烈影响,但PhyloBayes中实现的位点异质性混合模型避免了具有相似碱基组成和加速进化速率的不相关分类群的错误分组。通过观察变异性排序方法纳入rRNA基因并去除快速进化的位点以识别偏离平均速率的位点,改进了位点异质性模型下的系统发育推断,正确恢复了全变态类系统发育的大多数深层分支。我们建议,使用线粒体基因组数据来解决深层系统发育关系需要通过数据删除策略和使用位点异质性混合模型来评估替换饱和和组成偏差的潜在影响。我们的研究提出了一种在系统发育分析中如何使用密集采样的线粒体基因组数据的实用方法。