Jarquin Diego, Specht James, Lorenz Aaron
Department of Agronomy and Horticulture, University of Nebraska-Lincoln, Nebraska 68583-0915.
Department of Agronomy and Plant Genetics, University of Minnesota, St. Paul, Minnesota 55108
G3 (Bethesda). 2016 Aug 9;6(8):2329-41. doi: 10.1534/g3.116.031443.
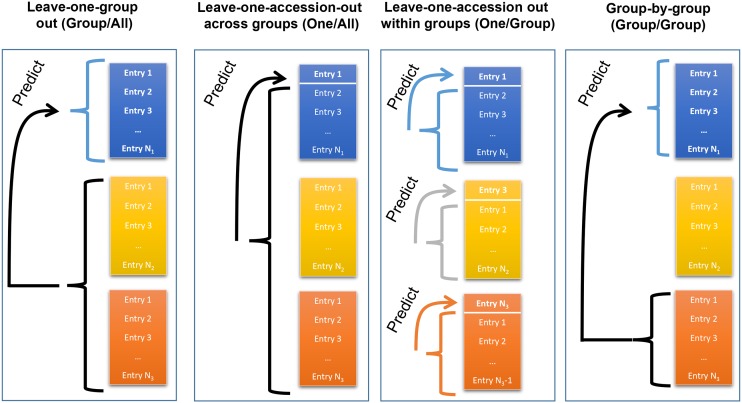
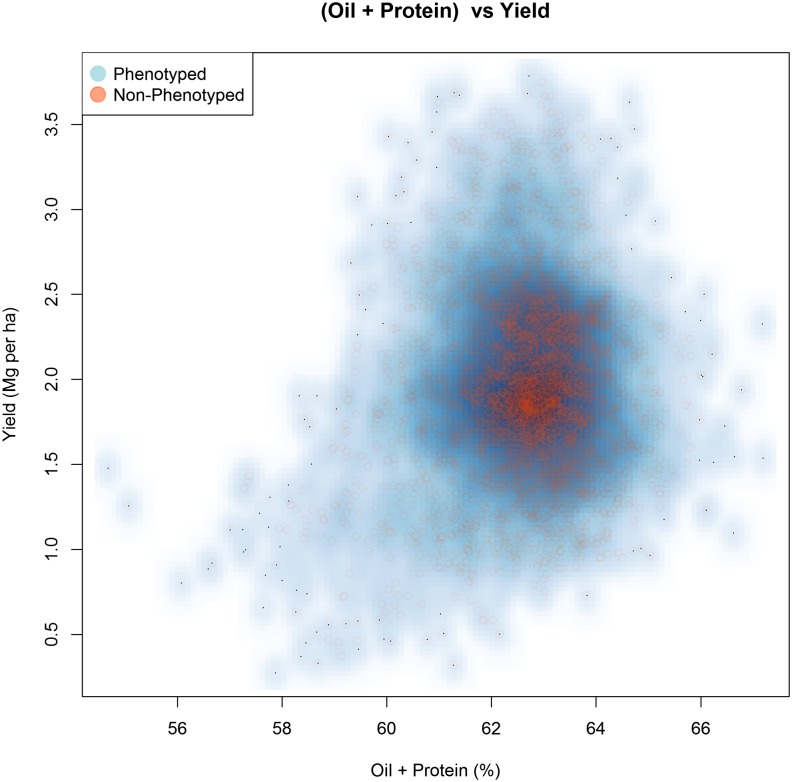
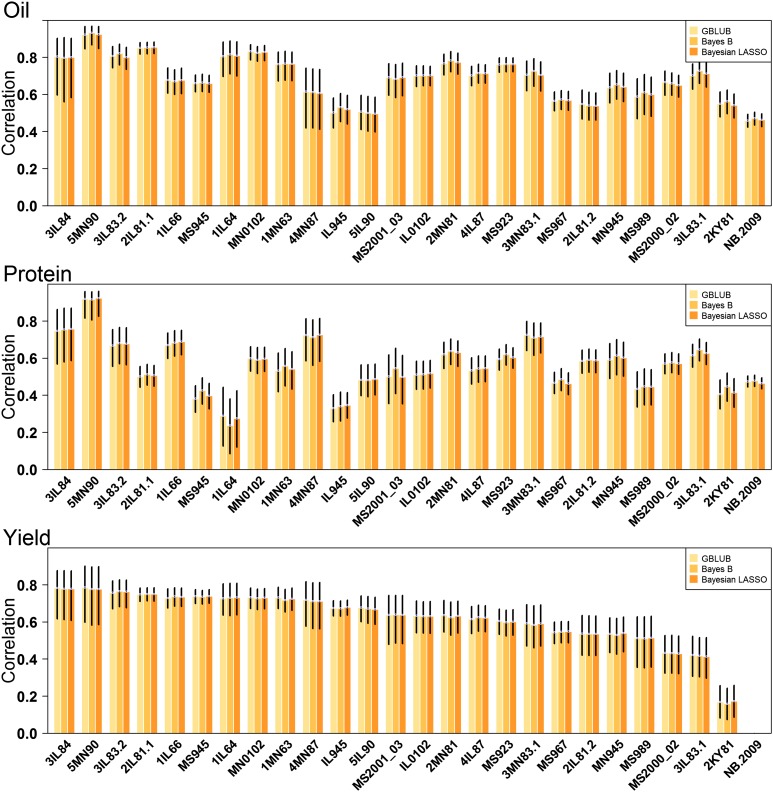
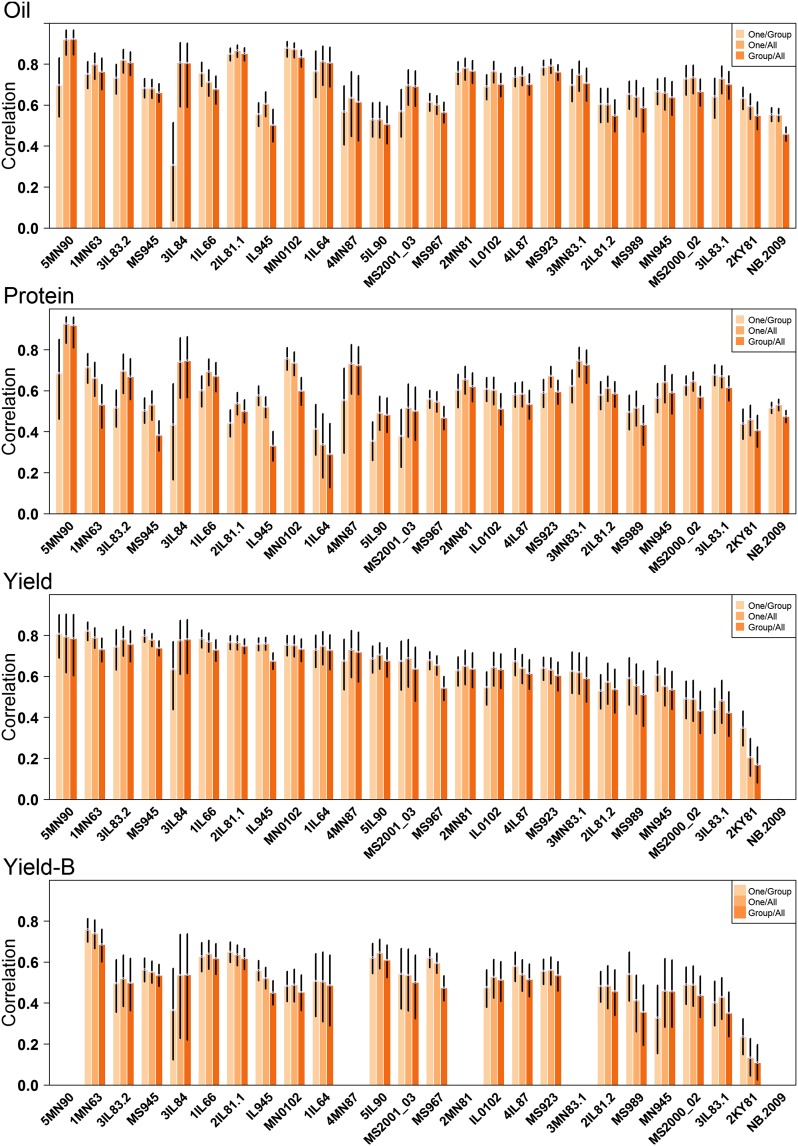
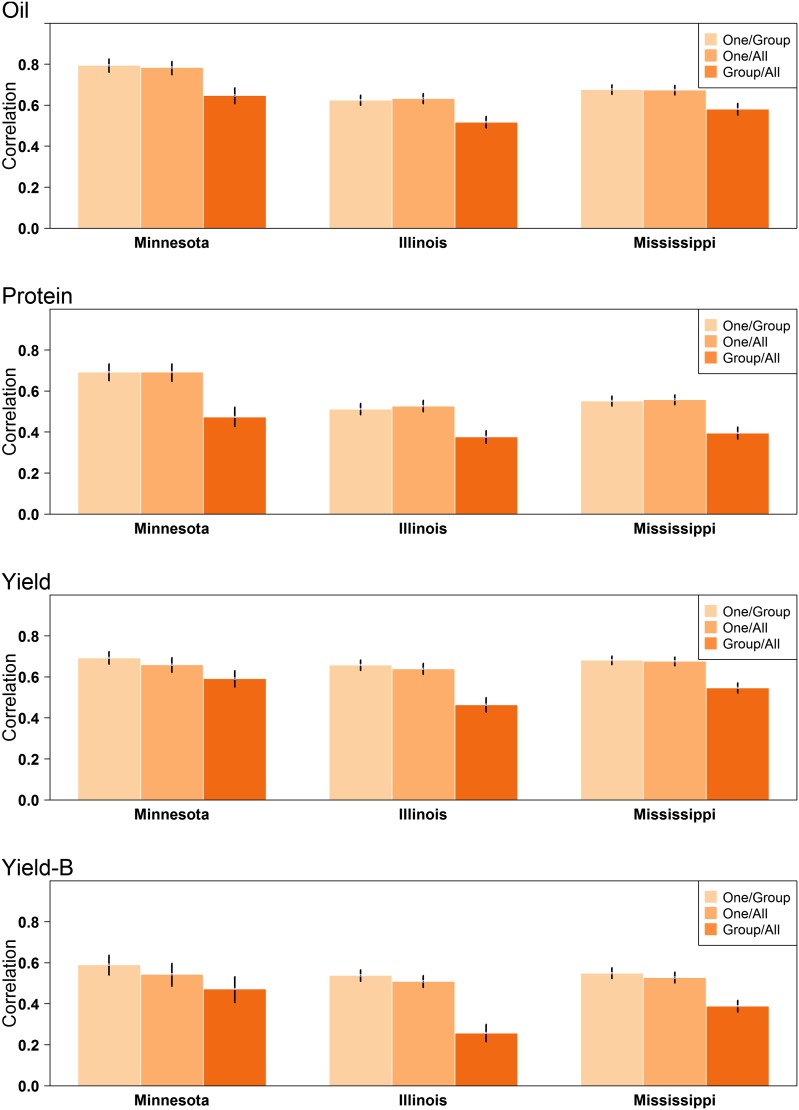
The identification and mobilization of useful genetic variation from germplasm banks for use in breeding programs is critical for future genetic gain and protection against crop pests. Plummeting costs of next-generation sequencing and genotyping is revolutionizing the way in which researchers and breeders interface with plant germplasm collections. An example of this is the high density genotyping of the entire USDA Soybean Germplasm Collection. We assessed the usefulness of 50K single nucleotide polymorphism data collected on 18,480 domesticated soybean (Glycine max) accessions and vast historical phenotypic data for developing genomic prediction models for protein, oil, and yield. Resulting genomic prediction models explained an appreciable amount of the variation in accession performance in independent validation trials, with correlations between predicted and observed reaching up to 0.92 for oil and protein and 0.79 for yield. The optimization of training set design was explored using a series of cross-validation schemes. It was found that the target population and environment need to be well represented in the training set. Second, genomic prediction training sets appear to be robust to the presence of data from diverse geographical locations and genetic clusters. This finding, however, depends on the influence of shattering and lodging, and may be specific to soybean with its presence of maturity groups. The distribution of 7608 nonphenotyped accessions was examined through the application of genomic prediction models. The distribution of predictions of phenotyped accessions was representative of the distribution of predictions for nonphenotyped accessions, with no nonphenotyped accessions being predicted to fall far outside the range of predictions of phenotyped accessions.
从种质库中识别和利用有用的遗传变异用于育种计划,对于未来的遗传增益和抵御作物病虫害至关重要。下一代测序和基因分型成本的大幅下降正在彻底改变研究人员和育种者与植物种质资源库交互的方式。美国农业部大豆种质资源库的全基因组高密度基因分型就是一个例子。我们评估了在18480份驯化大豆(Glycine max)种质上收集的50K单核苷酸多态性数据以及大量历史表型数据对于开发蛋白质、油脂和产量的基因组预测模型的有用性。由此得到的基因组预测模型在独立验证试验中解释了种质表现中相当一部分变异,预测值与观测值之间的相关性对于油脂和蛋白质高达0.92,对于产量为0.79。使用一系列交叉验证方案探索了训练集设计的优化。结果发现,训练集中需要很好地体现目标群体和环境。其次,基因组预测训练集对于来自不同地理位置和遗传簇的数据的存在似乎具有稳健性。然而,这一发现取决于裂荚和倒伏的影响,可能特定于具有成熟组的大豆。通过应用基因组预测模型检查了7608份未进行表型鉴定的种质的分布情况。已进行表型鉴定的种质的预测分布代表了未进行表型鉴定的种质的预测分布,没有未进行表型鉴定的种质被预测落在已进行表型鉴定的种质的预测范围之外。