Ngkelo Anta, Richart Adèle, Kirk Jonathan A, Bonnin Philippe, Vilar Jose, Lemitre Mathilde, Marck Pauline, Branchereau Maxime, Le Gall Sylvain, Renault Nisa, Guerin Coralie, Ranek Mark J, Kervadec Anaïs, Danelli Luca, Gautier Gregory, Blank Ulrich, Launay Pierre, Camerer Eric, Bruneval Patrick, Menasche Philippe, Heymes Christophe, Luche Elodie, Casteilla Louis, Cousin Béatrice, Rodewald Hans-Reimer, Kass David A, Silvestre Jean-Sébastien
Institut National de la Santé et de la Recherche Médicale (INSERM), UMRS-970, Centre de Recherche Cardiovasculaire, Université Paris Descartes, Sorbonne Paris Cité, F-75015 Paris, France.
Division of Cardiology, Johns Hopkins Medical Institutions, Baltimore, MD 212015.
J Exp Med. 2016 Jun 27;213(7):1353-74. doi: 10.1084/jem.20160081.
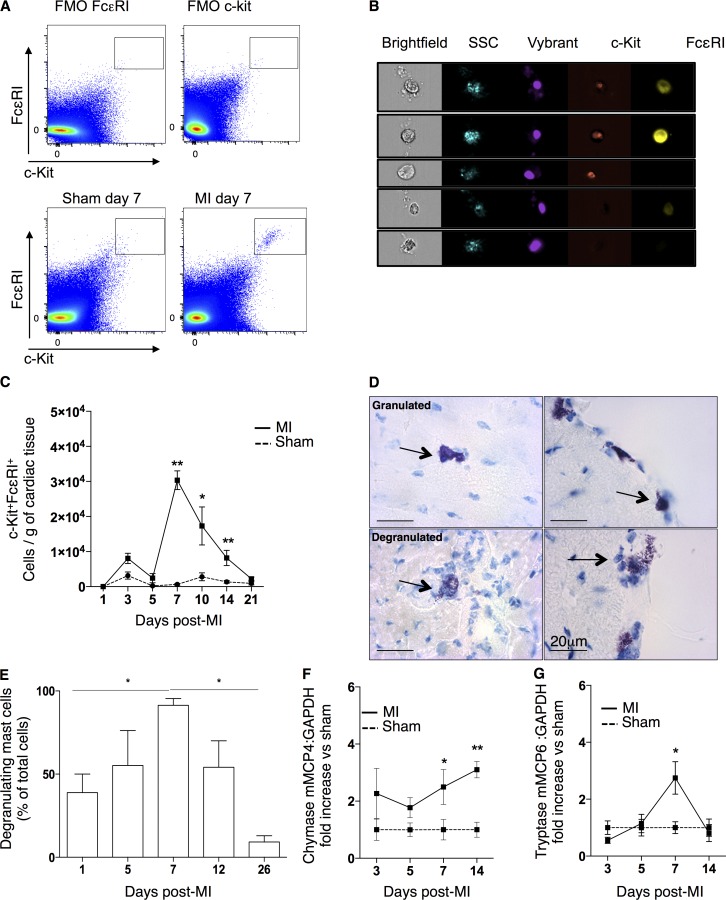
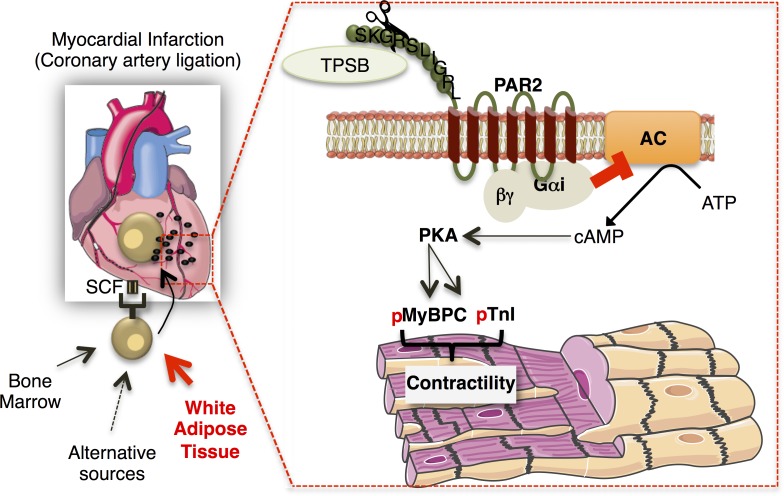
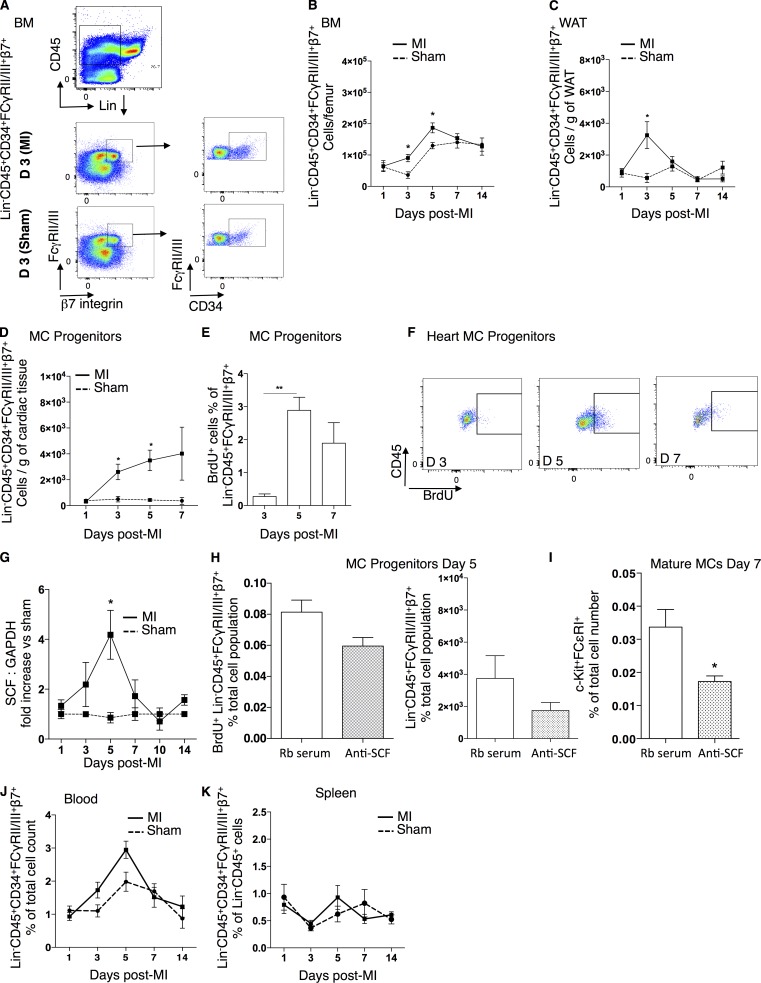
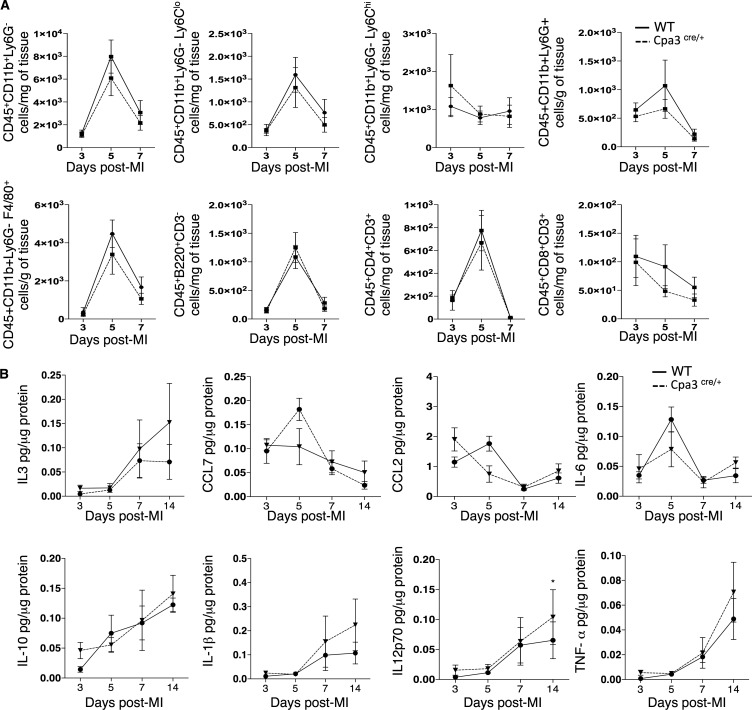
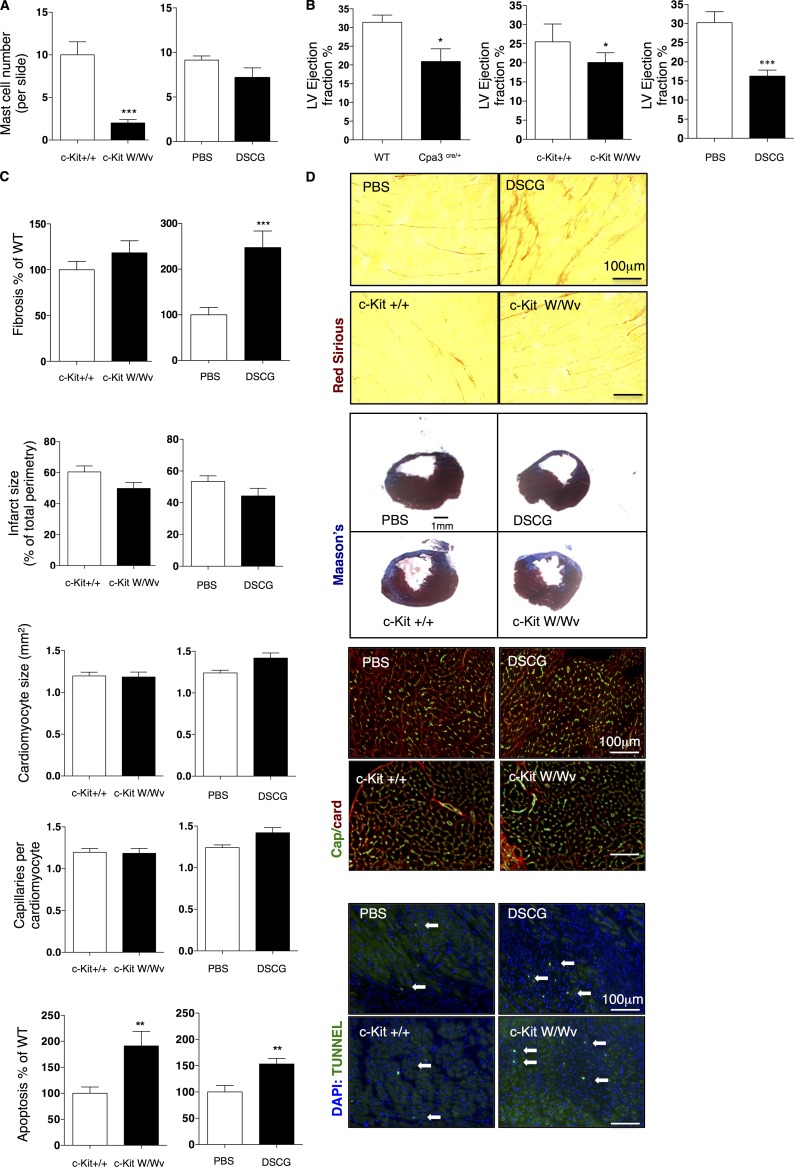
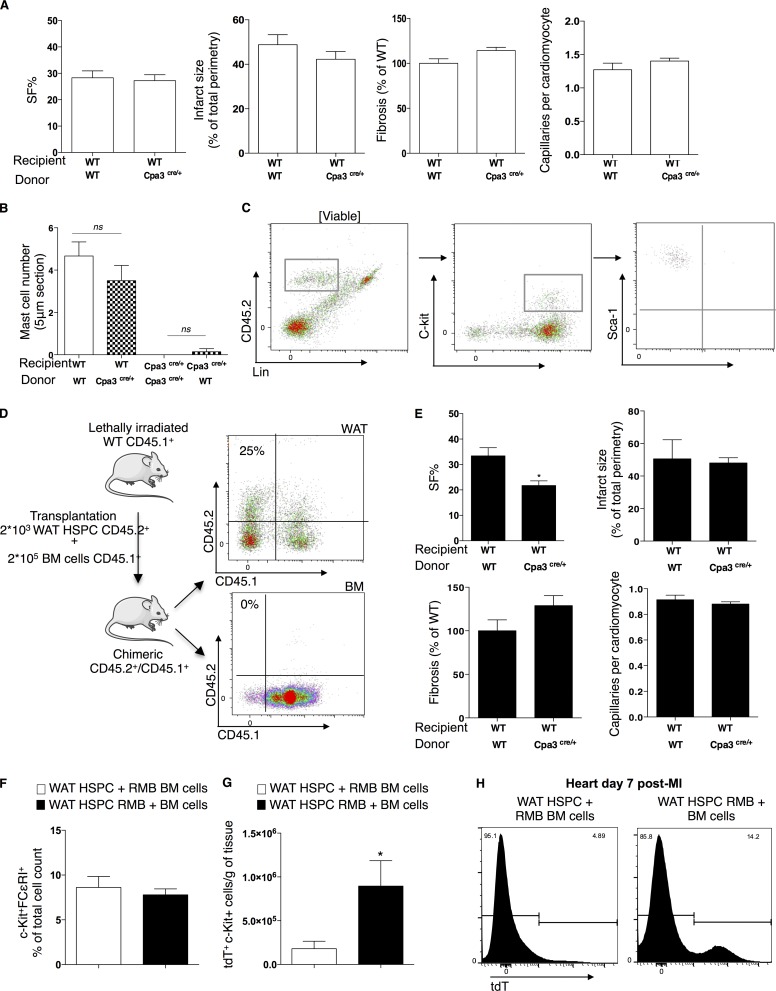
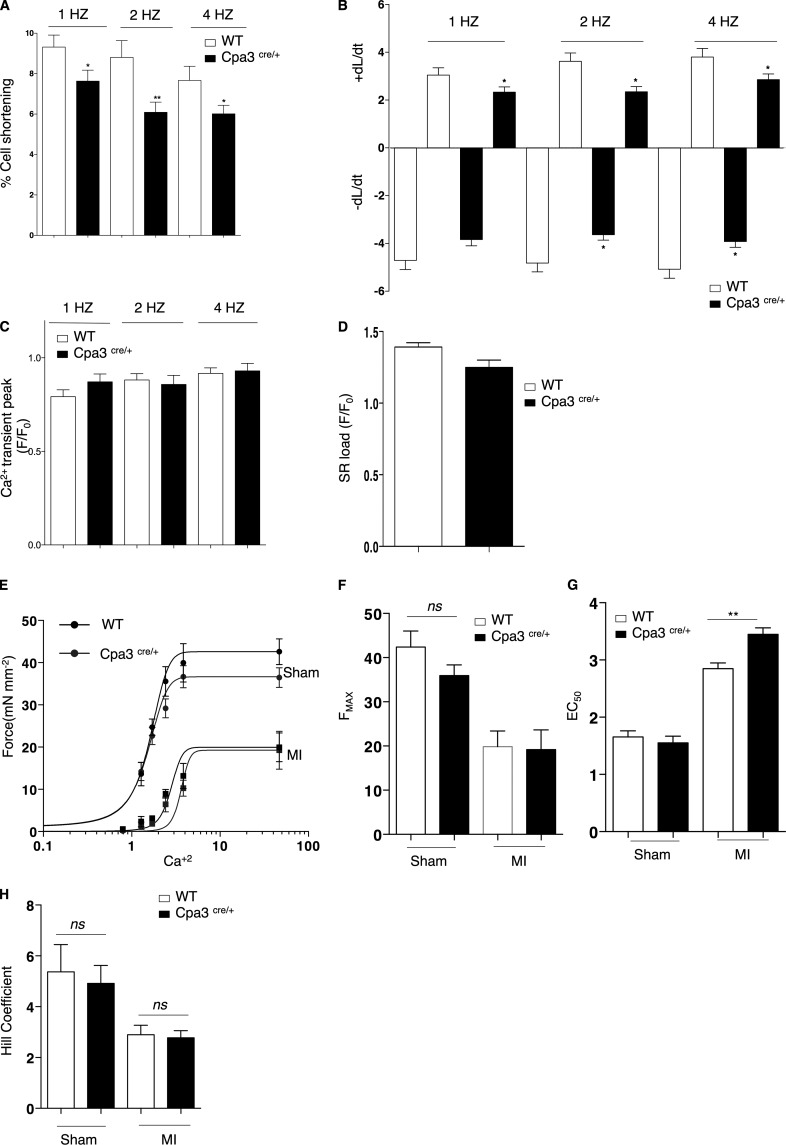
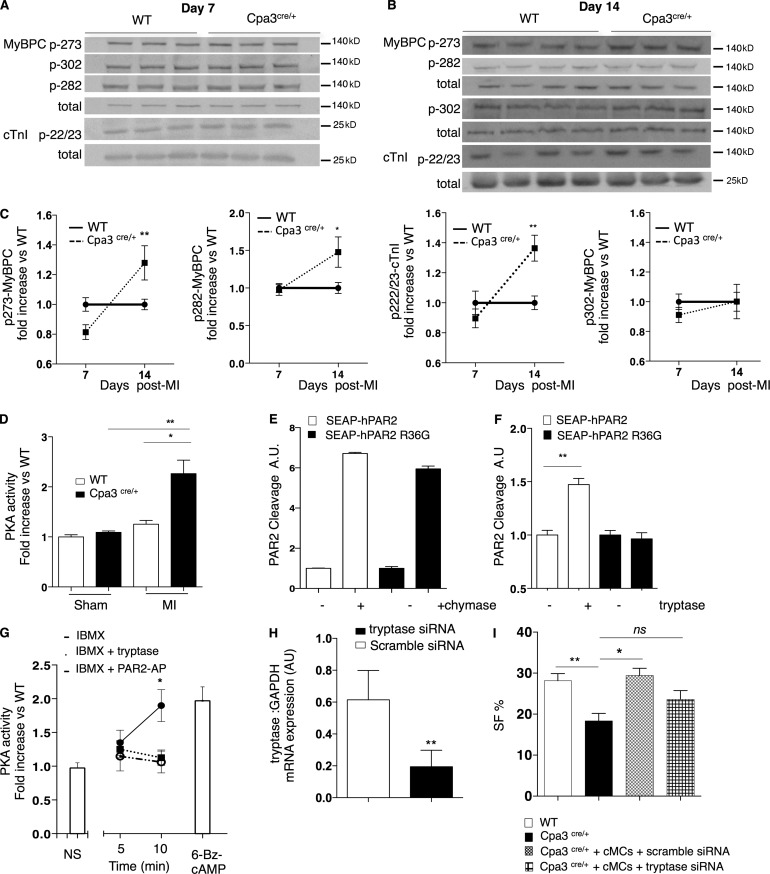
Acute myocardial infarction (MI) is a severe ischemic disease responsible for heart failure and sudden death. Inflammatory cells orchestrate postischemic cardiac remodeling after MI. Studies using mice with defective mast/stem cell growth factor receptor c-Kit have suggested key roles for mast cells (MCs) in postischemic cardiac remodeling. Because c-Kit mutations affect multiple cell types of both immune and nonimmune origin, we addressed the impact of MCs on cardiac function after MI, using the c-Kit-independent MC-deficient (Cpa3(Cre/+)) mice. In response to MI, MC progenitors originated primarily from white adipose tissue, infiltrated the heart, and differentiated into mature MCs. MC deficiency led to reduced postischemic cardiac function and depressed cardiomyocyte contractility caused by myofilament Ca(2+) desensitization. This effect correlated with increased protein kinase A (PKA) activity and hyperphosphorylation of its targets, troponin I and myosin-binding protein C. MC-specific tryptase was identified to regulate PKA activity in cardiomyocytes via protease-activated receptor 2 proteolysis. This work reveals a novel function for cardiac MCs modulating cardiomyocyte contractility via alteration of PKA-regulated force-Ca(2+) interactions in response to MI. Identification of this MC-cardiomyocyte cross-talk provides new insights on the cellular and molecular mechanisms regulating the cardiac contractile machinery and a novel platform for therapeutically addressable regulators.
急性心肌梗死(MI)是一种导致心力衰竭和猝死的严重缺血性疾病。炎症细胞在MI后协调缺血性心脏重塑。使用肥大/干细胞生长因子受体c-Kit缺陷小鼠的研究表明肥大细胞(MCs)在缺血性心脏重塑中起关键作用。由于c-Kit突变影响免疫和非免疫来源的多种细胞类型,我们使用不依赖c-Kit的MC缺陷(Cpa3(Cre/+))小鼠研究了MCs对MI后心脏功能的影响。对MI的反应中,MC祖细胞主要源自白色脂肪组织,浸润心脏并分化为成熟MCs。MC缺陷导致缺血后心脏功能降低以及由肌丝Ca(2+)脱敏引起的心肌细胞收缩力下降。这种效应与蛋白激酶A(PKA)活性增加及其靶点肌钙蛋白I和肌球蛋白结合蛋白C的过度磷酸化相关。已确定MC特异性组织蛋白酶通过蛋白酶激活受体2的蛋白水解作用调节心肌细胞中的PKA活性。这项工作揭示了心脏MCs的一种新功能,即通过改变PKA调节的力-Ca(2+)相互作用来调节心肌细胞收缩力以应对MI。这种MC-心肌细胞相互作用的鉴定为调节心脏收缩机制的细胞和分子机制提供了新见解,并为可治疗的调节因子提供了一个新平台。