Kuo Tzu-Hao, Li Kuo-Bin
Institute of Biomedical Informatics, National Yang-Ming University, Taipei 112, Taiwan.
Office of Information Management, National Yang-Ming University Hospital, Yilan 260, Taiwan.
Int J Mol Sci. 2016 Oct 26;17(11):1788. doi: 10.3390/ijms17111788.
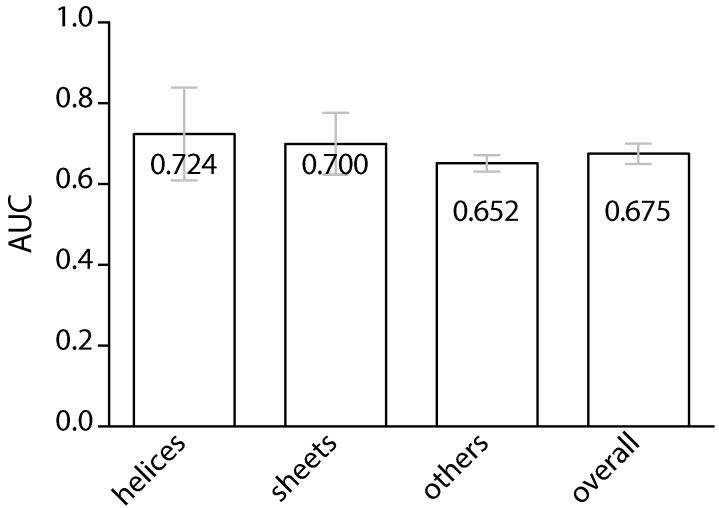
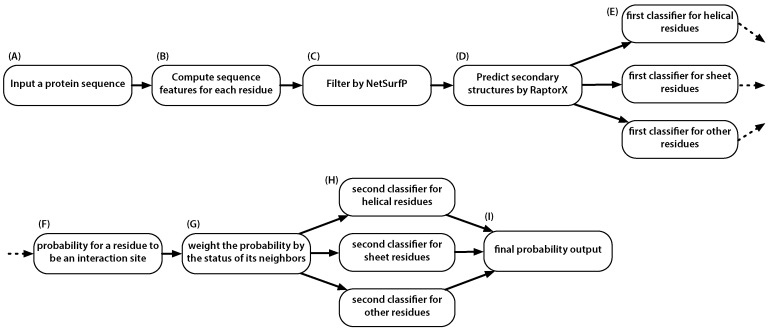
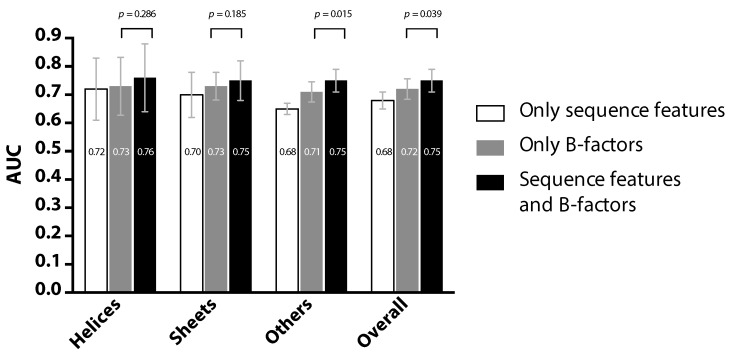
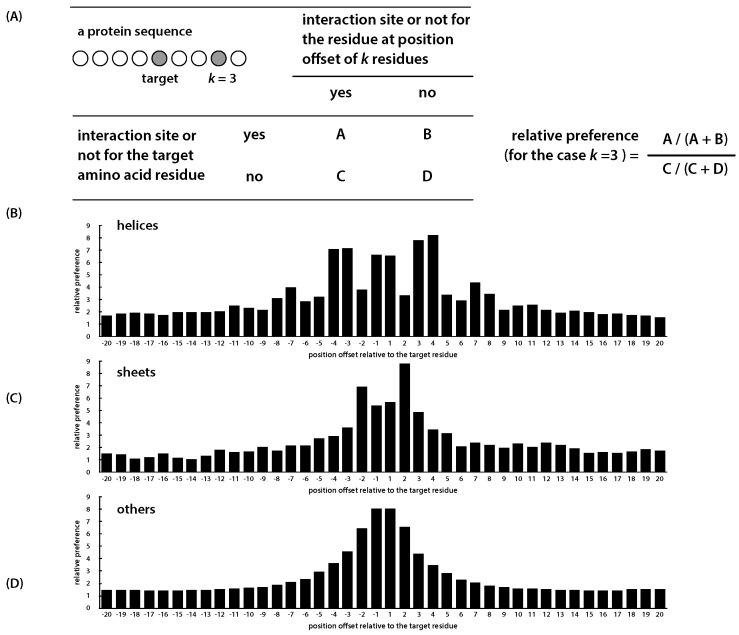
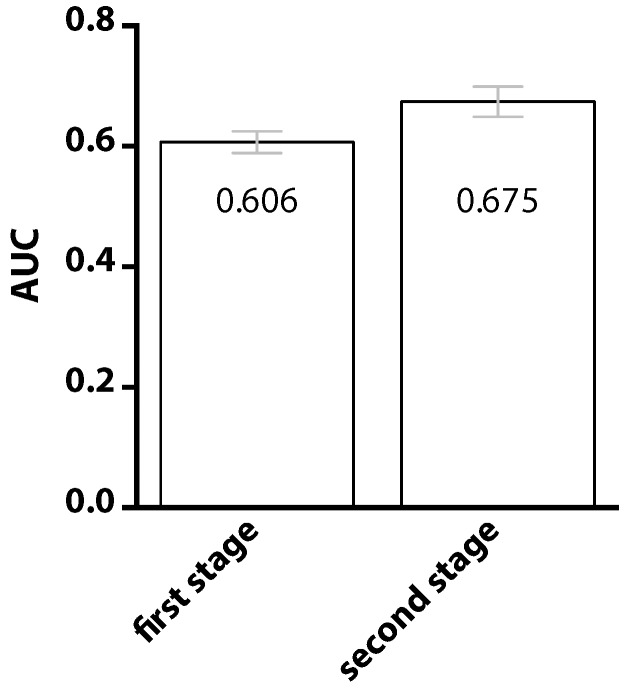
Information about the interface sites of Protein-Protein Interactions (PPIs) is useful for many biological research works. However, despite the advancement of experimental techniques, the identification of PPI sites still remains as a challenging task. Using a statistical learning technique, we proposed a computational tool for predicting PPI interaction sites. As an alternative to similar approaches requiring structural information, the proposed method takes all of the input from protein sequences. In addition to typical sequence features, our method takes into consideration that interaction sites are not randomly distributed over the protein sequence. We characterized this positional preference using protein complexes with known structures, proposed a numerical index to estimate the propensity and then incorporated the index into a learning system. The resulting predictor, without using structural information, yields an area under the ROC curve (AUC) of 0.675, recall of 0.597, precision of 0.311 and accuracy of 0.583 on a ten-fold cross-validation experiment. This performance is comparable to the previous approach in which structural information was used. Upon introducing the B-factor data to our predictor, we demonstrated that the AUC can be further improved to 0.750. The tool is accessible at http://bsaltools.ym.edu.tw/predppis.
蛋白质-蛋白质相互作用(PPI)界面位点的信息对许多生物学研究工作都很有用。然而,尽管实验技术不断进步,但PPI位点的识别仍然是一项具有挑战性的任务。我们使用统计学习技术,提出了一种预测PPI相互作用位点的计算工具。作为需要结构信息的类似方法的替代方案,该方法从蛋白质序列获取所有输入。除了典型的序列特征外,我们的方法还考虑到相互作用位点并非随机分布在蛋白质序列上。我们利用具有已知结构的蛋白质复合物来表征这种位置偏好,提出了一个数值指标来估计倾向,然后将该指标纳入学习系统。在十折交叉验证实验中,所得的预测器在不使用结构信息的情况下,ROC曲线下面积(AUC)为0.675,召回率为0.597,精确率为0.311,准确率为0.583。这一性能与之前使用结构信息的方法相当。在将B因子数据引入我们的预测器后,我们证明AUC可以进一步提高到0.750。该工具可在http://bsaltools.ym.edu.tw/predppis获取。