Maurer A, Sannemann W, Léon J, Pillen K
Institute of Agricultural and Nutritional Sciences, Martin Luther University Halle-Wittenberg, Halle, Germany.
Institute for Crop Science and Resource Conservation, University Bonn, Bonn, Germany.
Heredity (Edinb). 2017 May;118(5):477-485. doi: 10.1038/hdy.2016.121. Epub 2016 Dec 14.
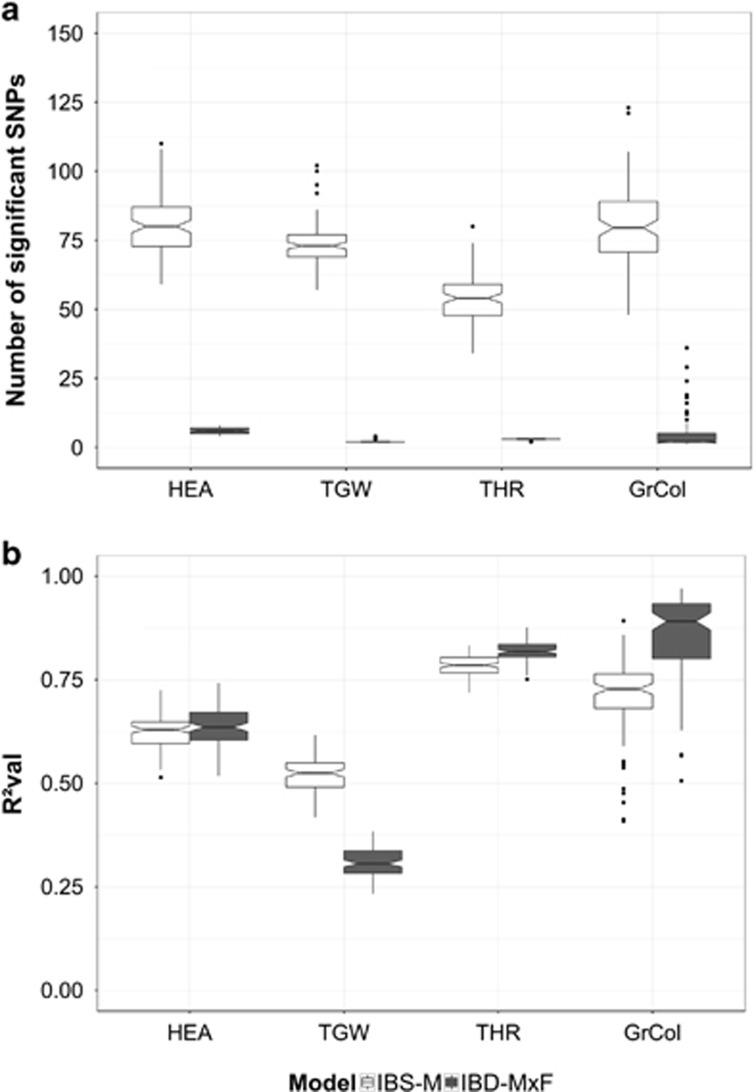
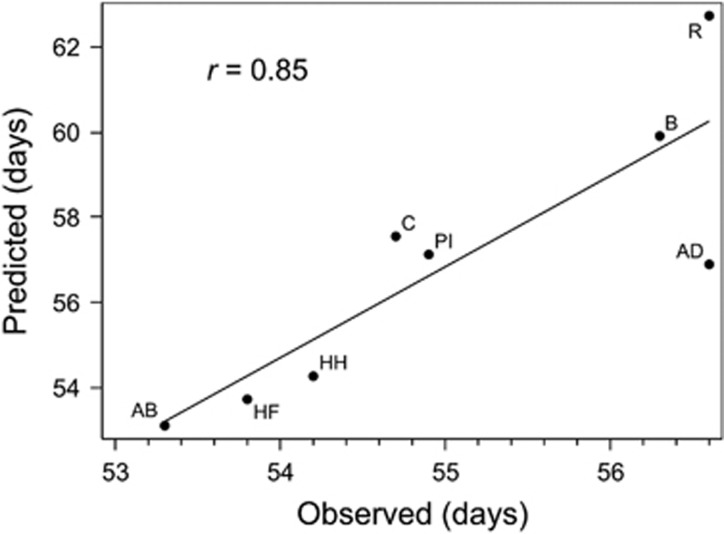
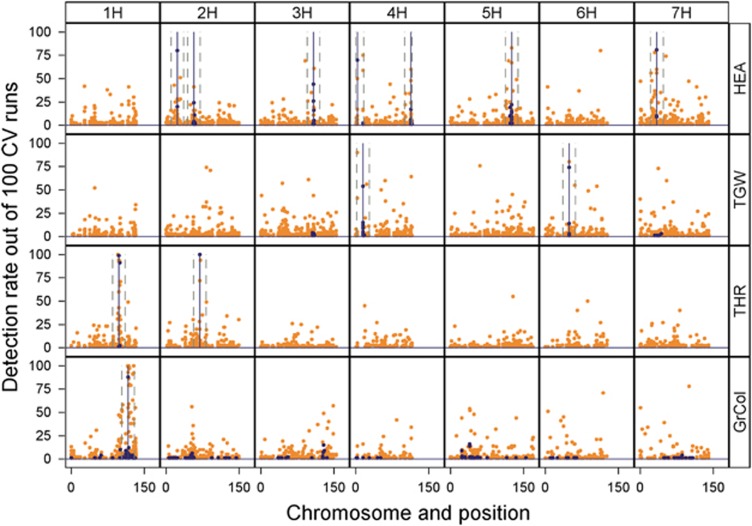
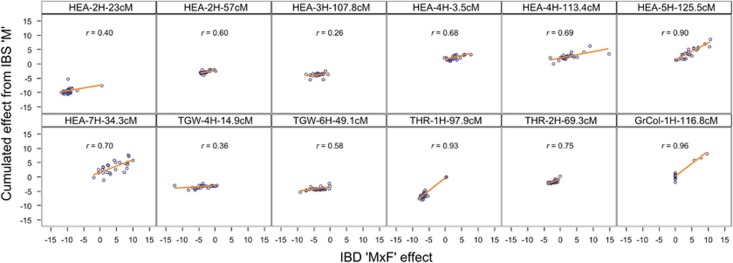
The emergence of multiparental mapping populations enabled plant geneticists to gain deeper insights into the genetic architecture of major agronomic traits and to map quantitative trait loci (QTLs) controlling the expression of these traits. Although the investigated mapping populations are similar, one open question is whether genotype data should be modelled as identical by state (IBS) or identical by descent (IBD). Whereas IBS simply makes use of raw genotype scores to distinguish alleles, IBD data are derived from parental offspring information. We report on comparing IBS and IBD by applying two multiple regression models on four traits studied in the barley nested association mapping (NAM) population HEB-25. We observed that modelling parent-specific IBD genotypes produced a lower number of significant QTLs with increased prediction abilities compared with modelling IBS genotypes. However, at lower trait heritabilities the IBS model produced higher prediction abilities. We developed a method to estimate multiallelic QTL effects in multiparental populations from simple biallelic IBS data. This method is based on cumulating IBS-derived single-nucleotide polymorphism (SNP) effect estimates in a defined genetic region surrounding a QTL. Comparing the resulting parent-specific QTL effects with those obtained from IBD approaches revealed high accordance that could be confirmed through simulations. The method turned out to be also applicable to a barley multiparent advanced generation inter-cross (MAGIC) population. The 'cumulation method' represents a universal approach to differentiate parent-specific QTL effects in multiparental populations, even if no IBD information is available. In future, the method could further benefit from the availability of much denser SNP maps.
多亲本表型群体的出现使植物遗传学家能够更深入地了解主要农艺性状的遗传结构,并定位控制这些性状表达的数量性状位点(QTL)。尽管所研究的表型群体相似,但一个悬而未决的问题是,基因型数据应建模为状态相同(IBS)还是血缘相同(IBD)。IBS只是简单地利用原始基因型得分来区分等位基因,而IBD数据则来自亲本后代信息。我们通过在大麦巢式关联作图(NAM)群体HEB - 25中研究的四个性状上应用两个多元回归模型来比较IBS和IBD。我们观察到,与对IBS基因型进行建模相比,对亲本特异性IBD基因型进行建模产生的显著QTL数量更少,但预测能力有所提高。然而,在较低的性状遗传力下,IBS模型具有更高的预测能力。我们开发了一种方法,可从简单的双等位基因IBS数据估计多亲本表型群体中的多等位基因QTL效应。该方法基于在QTL周围定义的遗传区域中累积IBS衍生的单核苷酸多态性(SNP)效应估计值。将所得的亲本特异性QTL效应与从IBD方法获得的效应进行比较,发现高度一致,这可以通过模拟得到证实。该方法也适用于大麦多亲高代杂交(MAGIC)群体。“累积法”代表了一种通用方法,即使没有IBD信息,也能区分多亲本表型群体中亲本特异性的QTL效应。未来,该方法可能会从更密集的SNP图谱中进一步受益。