Center for Computational Biology and Department of Molecular Biosciences, University of Kansas, Lawrence, Kansas 66047, USA.
J Chem Phys. 2018 Aug 21;149(7):072308. doi: 10.1063/1.5024217.
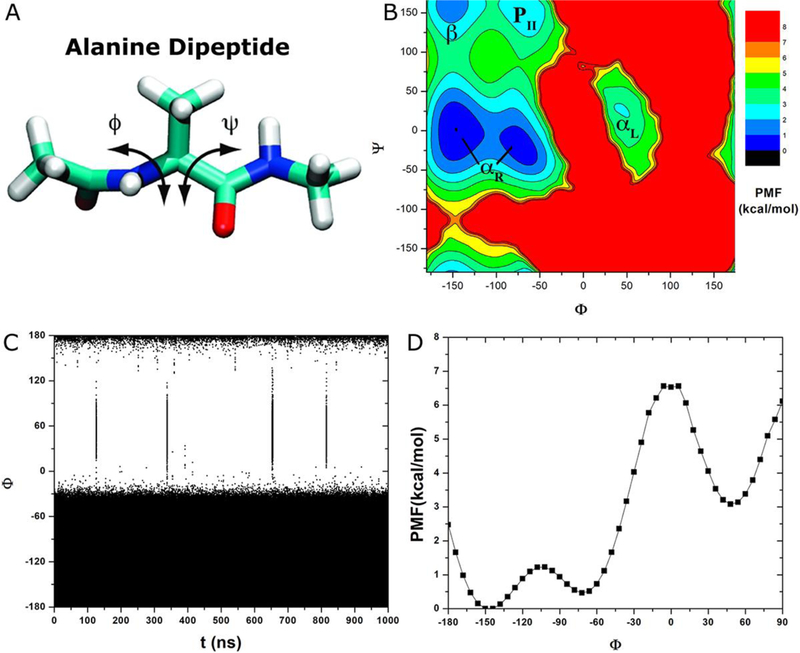
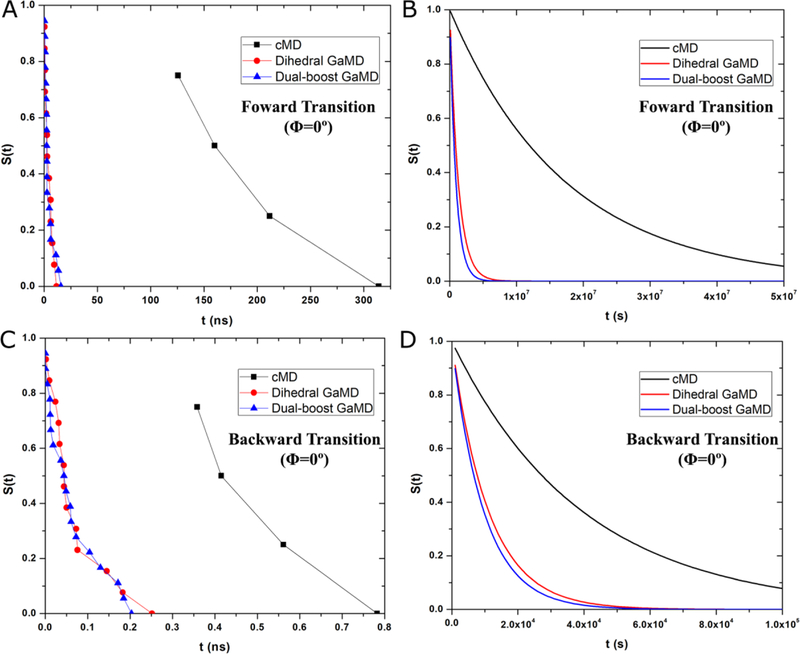
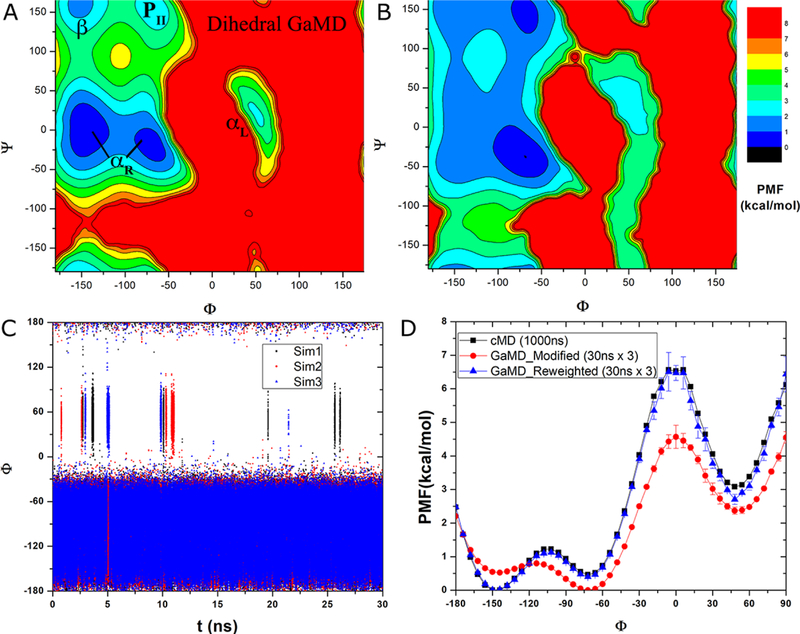
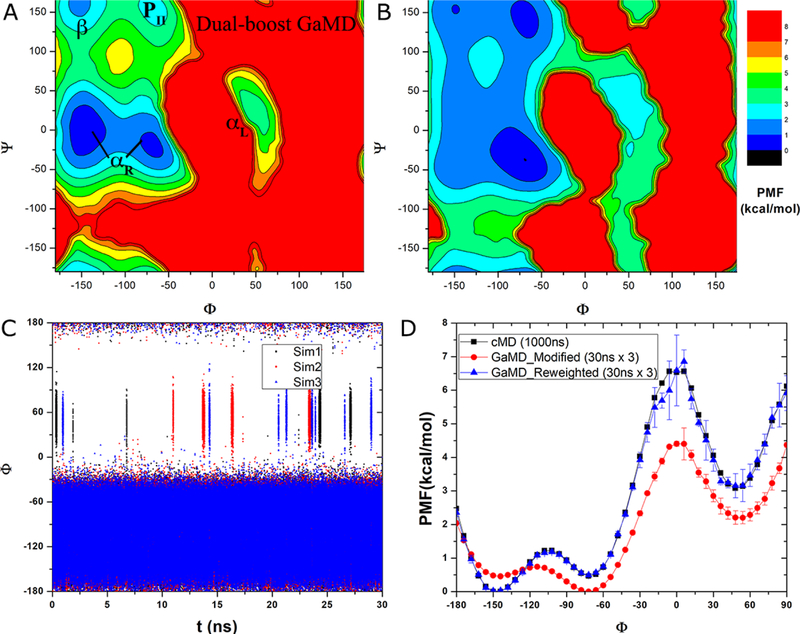
Recent studies demonstrated that Gaussian accelerated molecular dynamics (GaMD) is a robust computational technique, which provides simultaneous unconstrained enhanced sampling and free energy calculations of biomolecules. However, the exact acceleration of biomolecular dynamics or speedup of kinetic rates in GaMD simulations and, more broadly, in enhanced sampling methods, remains a challenging task to be determined. Here, the GaMD acceleration is examined using alanine dipeptide in explicit solvent as a biomolecular model system. Relative to long conventional molecular dynamics simulation, GaMD simulations exhibited ∼36-67 times speedup for sampling of the backbone dihedral transitions. The acceleration depended on level of the GaMD boost potential. Furthermore, Kramers' rate theory was applied to estimate GaMD acceleration using simulation-derived diffusion coefficients, curvatures and barriers of free energy profiles. In most cases, the calculations also showed significant speedup of dihedral transitions in GaMD, although the GaMD acceleration factors tended to be underestimated by ∼3-96 fold. Because greater boost potential can be applied in GaMD simulations of systems with increased sizes, which potentially leads to higher acceleration, it is subject to future studies on accelerating the dynamics and recovering kinetic rates of larger biomolecules such as proteins and protein-protein/nucleic acid complexes.
最近的研究表明,高斯加速分子动力学(GaMD)是一种稳健的计算技术,可同时对生物分子进行无约束增强采样和自由能计算。然而,GaMD 模拟中的生物分子动力学的精确加速或动力学速率的加速,更广泛地说,在增强采样方法中,仍然是一个有待确定的具有挑战性的任务。在这里,使用明溶剂中的丙氨酸二肽作为生物分子模型系统来检查 GaMD 加速。与传统的长分子动力学模拟相比,GaMD 模拟在采样主链二面角转变方面表现出约 36-67 倍的加速。这种加速取决于 GaMD 提升势的水平。此外,应用 Kramer 速率理论,使用模拟衍生的扩散系数、自由能曲线的曲率和势垒来估计 GaMD 加速。在大多数情况下,计算还表明 GaMD 中的二面角转变明显加速,尽管 GaMD 加速因子往往被低估了约 3-96 倍。因为可以在具有更大尺寸的系统的 GaMD 模拟中应用更大的提升势,这可能导致更高的加速,因此它是未来研究更大的生物分子(如蛋白质和蛋白质/核酸复合物)的动力学加速和恢复动力学速率的主题。