Mahapatra Saugata, Gallaher Brandi, Smith Sydni Caet, Graham Joseph G, Voth Daniel E, Shaw Edward I
Department of Microbiology and Molecular genetics, Oklahoma State University Stillwater, OK, USA.
Department of Microbiology and Immunology, University of Arkansas for Medical Sciences (UAMS) Little Rock, AR, USA.
Front Cell Infect Microbiol. 2016 Dec 19;6:188. doi: 10.3389/fcimb.2016.00188. eCollection 2016.
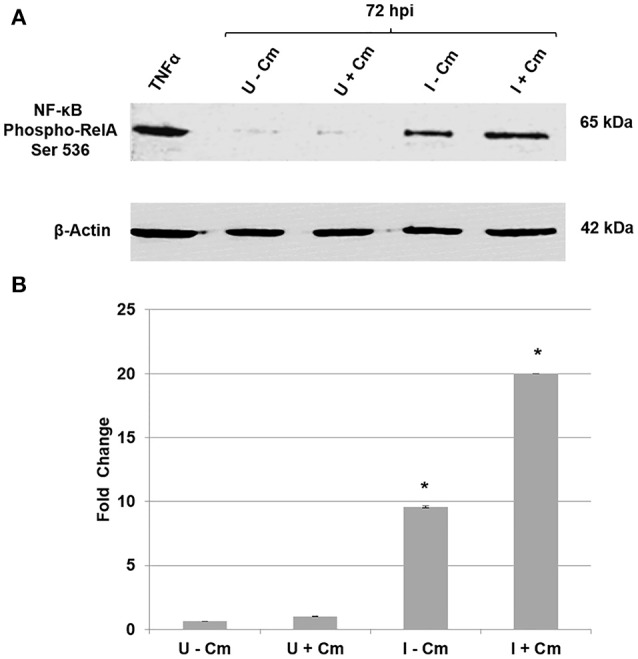
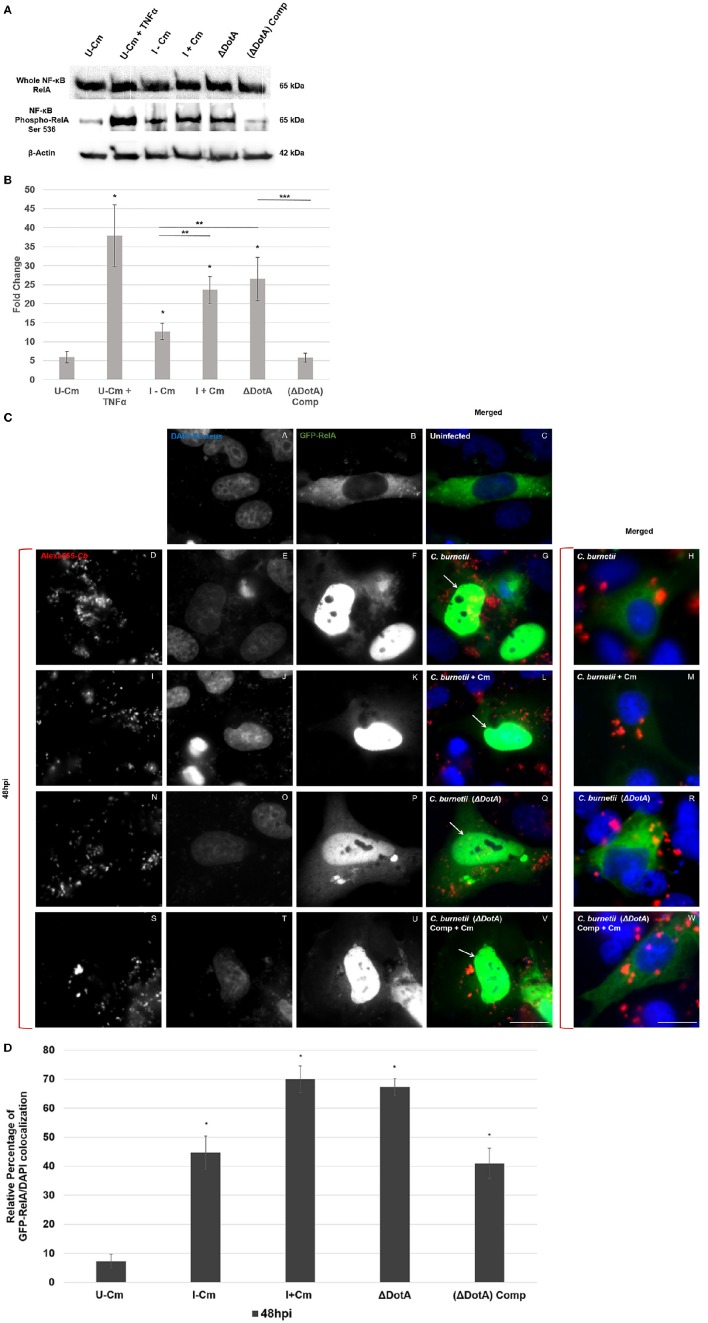
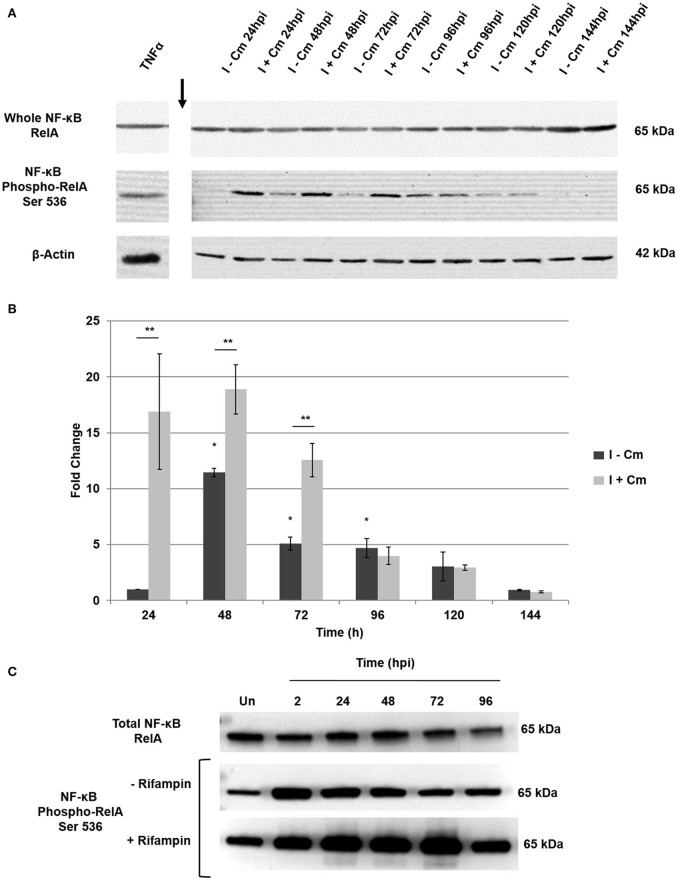
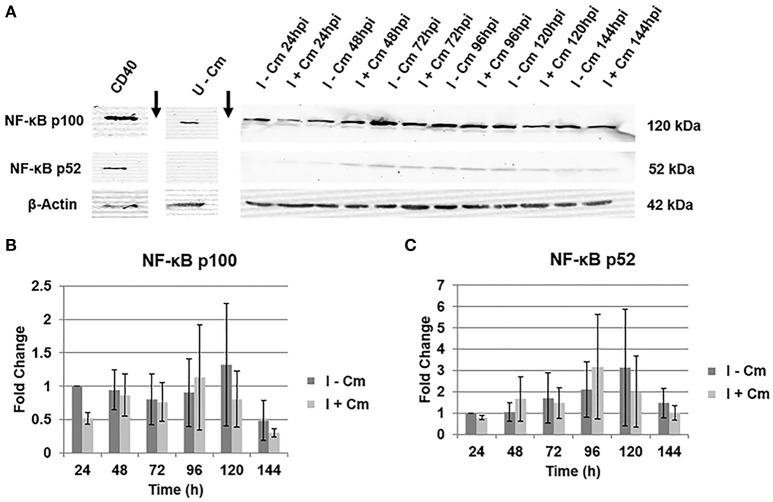
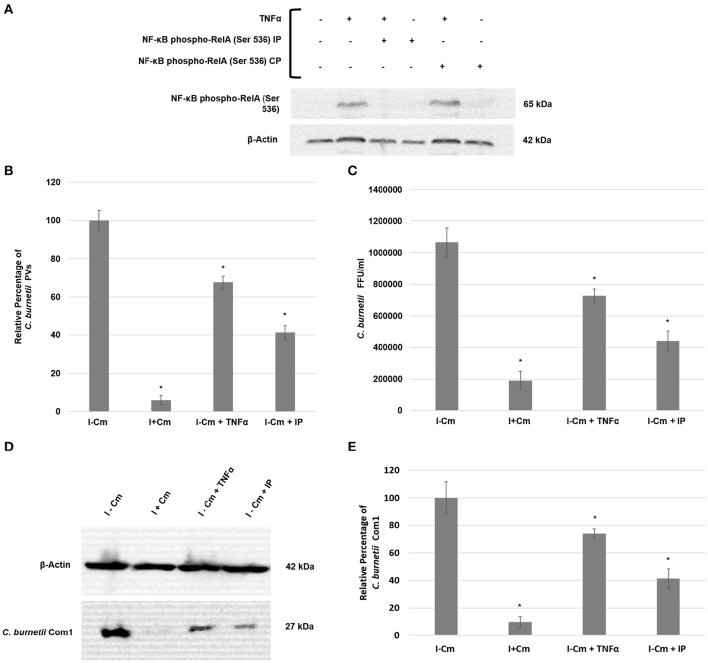
is the causative agent of Q fever and an obligate intracellular pathogen in nature that survives and grows in a parasitophorous vacuole (PV) within eukaryotic host cells. promotes intracellular survival by subverting apoptotic and pro-inflammatory signaling pathways that are typically regulated by nuclear transcription factor-κB (NF-κB). We and others have demonstrated that NMII proteins inhibit expression of pro-inflammatory cytokines and induce expression of anti-apoptotic genes during infection. Here, we demonstrate that promotes intracellular survival by modulating NF-κB subunit p65 (RelA) phosphorylation, and thus activation, in a Type Four B Secretion System (T4BSS)-dependent manner. Immunoblot analysis of RelA phosphorylated at serine-536 demonstrated that increases NF-κB activation via the canonical pathway. However, RelA phosphorylation levels were even higher in infected cells where bacterial protein or mRNA synthesis was inhibited. Importantly, we demonstrate that inhibition of RelA phosphorylation impairs PV formation and growth. We found that a T4BSS-defective mutant (CbΔA) elicited phosphorylated RelA levels similar to those of wild type infection treated with Chloramphenicol. Moreover, cells infected with CbΔA or wild type treated with Chloramphenicol showed similar levels of GFP-RelA nuclear localization, and significantly increased localization compared to wild type infection. These data indicate that without protein synthesis and a functional T4BSS, is unable to modulate NF-κB activation, which is crucial for optimal intracellular growth.
是Q热的病原体,本质上是一种专性细胞内病原体,在真核宿主细胞内的寄生泡(PV)中存活和生长。通过颠覆通常由核转录因子-κB(NF-κB)调节的凋亡和促炎信号通路来促进细胞内存活。我们和其他人已经证明,NMII蛋白在感染期间抑制促炎细胞因子的表达并诱导抗凋亡基因的表达。在这里,我们证明通过以IV型B分泌系统(T4BSS)依赖的方式调节NF-κB亚基p65(RelA)的磷酸化,从而激活,来促进细胞内存活。对丝氨酸-536处磷酸化的RelA进行免疫印迹分析表明,通过经典途径增加NF-κB的激活。然而,在细菌蛋白或mRNA合成受到抑制的感染细胞中,RelA的磷酸化水平甚至更高。重要的是,我们证明抑制RelA磷酸化会损害PV形成和生长。我们发现,一个T4BSS缺陷突变体(CbΔA)引发的磷酸化RelA水平与用氯霉素处理的野生型感染相似。此外,用氯霉素处理的感染CbΔA或野生型的细胞显示出相似水平的GFP-RelA核定位,并且与野生型感染相比,定位显著增加。这些数据表明,没有蛋白质合成和功能性T4BSS,就无法调节NF-κB的激活,而NF-κB的激活对于最佳细胞内生长至关重要。