Sauerwald Natalie, Zhang She, Kingsford Carl, Bahar Ivet
Computational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA.
Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USA.
Nucleic Acids Res. 2017 Apr 20;45(7):3663-3673. doi: 10.1093/nar/gkx172.
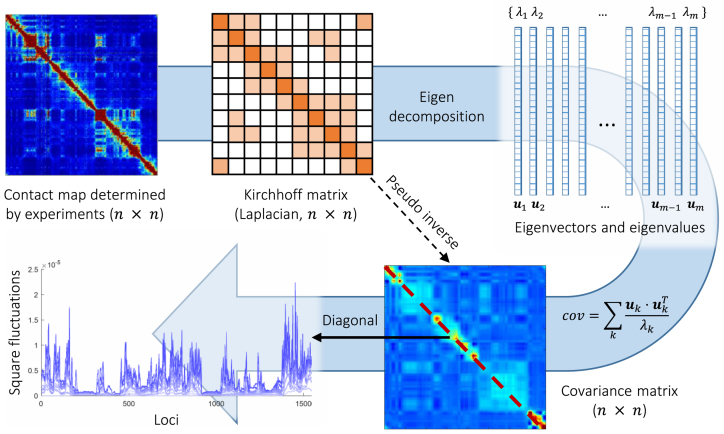
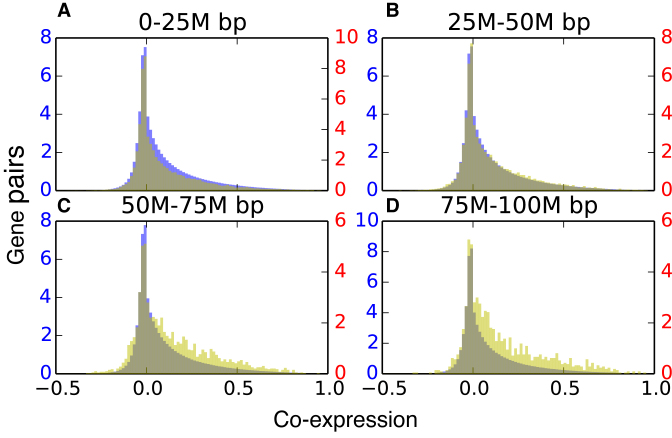
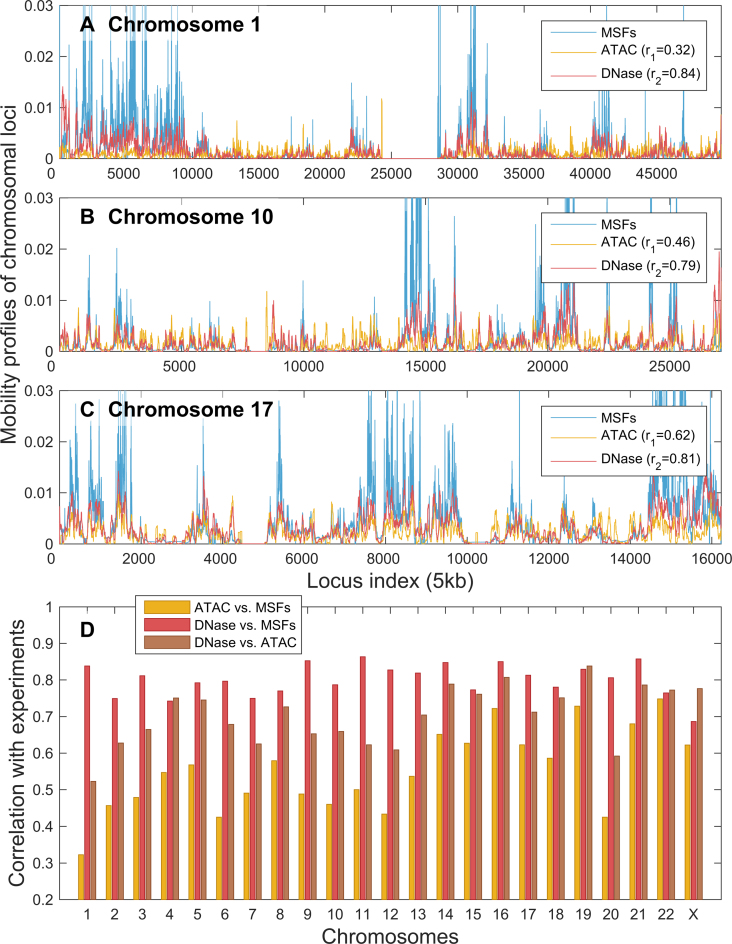
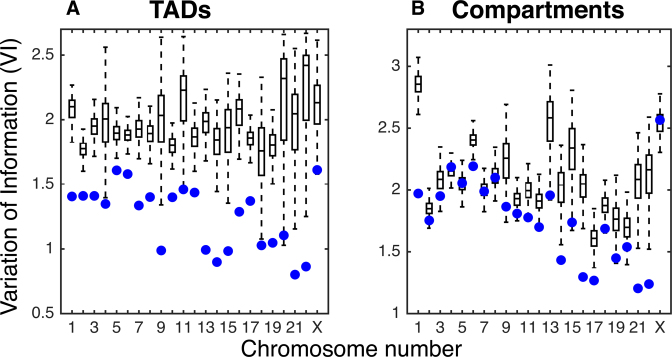
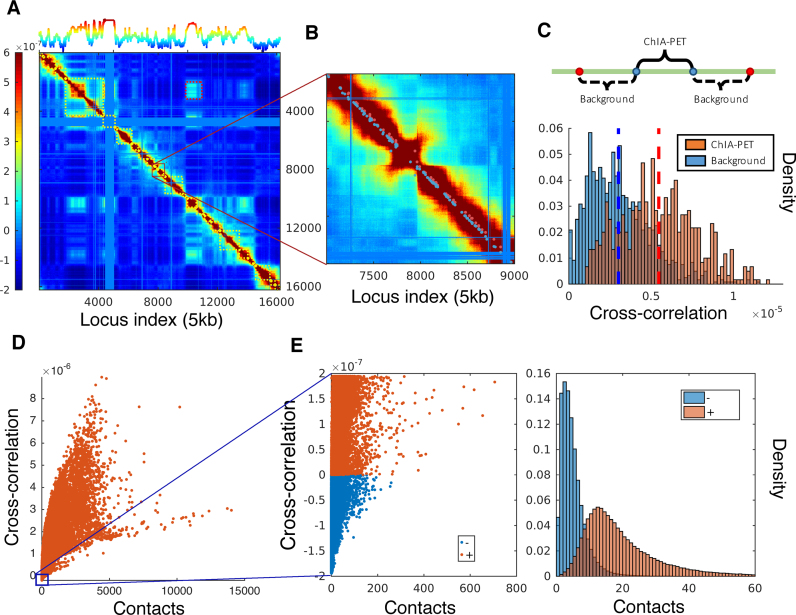
Understanding the three-dimensional (3D) architecture of chromatin and its relation to gene expression and regulation is fundamental to understanding how the genome functions. Advances in Hi-C technology now permit us to study 3D genome organization, but we still lack an understanding of the structural dynamics of chromosomes. The dynamic couplings between regions separated by large genomic distances (>50 Mb) have yet to be characterized. We adapted a well-established protein-modeling framework, the Gaussian Network Model (GNM), to model chromatin dynamics using Hi-C data. We show that the GNM can identify spatial couplings at multiple scales: it can quantify the correlated fluctuations in the positions of gene loci, find large genomic compartments and smaller topologically-associating domains (TADs) that undergo en bloc movements, and identify dynamically coupled distal regions along the chromosomes. We show that the predictions of the GNM correlate well with genome-wide experimental measurements. We use the GNM to identify novel cross-correlated distal domains (CCDDs) representing pairs of regions distinguished by their long-range dynamic coupling and show that CCDDs are associated with increased gene co-expression. Together, these results show that GNM provides a mathematically well-founded unified framework for modeling chromatin dynamics and assessing the structural basis of genome-wide observations.
理解染色质的三维(3D)结构及其与基因表达和调控的关系是理解基因组如何发挥功能的基础。Hi-C技术的进展使我们能够研究三维基因组组织,但我们仍缺乏对染色体结构动力学的理解。由大基因组距离(>50 Mb)分隔的区域之间的动态耦合尚未得到表征。我们采用了一个成熟的蛋白质建模框架,即高斯网络模型(GNM),利用Hi-C数据对染色质动力学进行建模。我们表明,GNM可以在多个尺度上识别空间耦合:它可以量化基因座位置的相关波动,找到经历整体移动的大基因组区域和较小的拓扑相关结构域(TAD),并识别沿染色体动态耦合的远端区域。我们表明,GNM的预测与全基因组实验测量结果密切相关。我们使用GNM来识别代表通过长程动态耦合区分的区域对的新型交叉相关远端结构域(CCDD),并表明CCDD与增加的基因共表达相关。总之,这些结果表明,GNM为建模染色质动力学和评估全基因组观察的结构基础提供了一个数学上有充分依据的统一框架。